

Heart failure (HF) is a clinical syndrome in which the heart is not able to meet the metabolic demands of the body due to a structural and or functional cardiac abnormality, leading to low cardiac output, elevated ventricular filling pressure, or both. Acute heart failure includes both new-onset heart failure and acute decompensated heart failure (an exacerbation of chronic heart failure). In both, there are progressive signs or symptoms of cardiac congestion, low output, or both.. Timely medical intervention, directed at reducing symptoms and managing the cause, is necessary.
(See also Overview of Heart Failure.)
Acute heart failure refers not only to the timing of clinical presentation within the overall disease course, but the importance of timely medical intervention (1). It may include new-onset (de novo) heart failure or acute decompensation of previously diagnosed heart failure. Acute heart failure is classified phenotypically and further risk-stratified based on the overall severity of illness, alteration in perfusion ("warm" versus "cold"), the presence of congestion ("dry" versus "wet"), and other details of clinical presentation (2, 3).
General references
1. Mebazaa A, Yilmaz MB, Levy P, et al. Recommendations on pre-hospital & early hospital management of acute heart failure: a consensus paper from the Heart Failure Association of the European Society of Cardiology, the European Society of Emergency Medicine and the Society of Academic Emergency Medicine. Eur J Heart Fail. 2015;17(6):544-558. doi:10.1002/ejhf.289
2. Arrigo M, Jessup M, Mullens W, et al. Acute heart failure. Nat Rev Dis Primers. 2020;6(1):16. Published 2020 Mar 5. doi:10.1038/s41572-020-0151-7
3. Hollenberg SM, Warner Stevenson L, Ahmad T, et al. 2019 ACC Expert Consensus Decision Pathway on Risk Assessment, Management, and Clinical Trajectory of Patients Hospitalized With Heart Failure: A Report of the American College of Cardiology Solution Set Oversight Committee [published correction appears in J Am Coll Cardiol. 2020 Jan 7;75(1):132. doi: 10.1016/j.jacc.2019.11.020.]. J Am Coll Cardiol. 2019;74(15):1966-2011. doi:10.1016/j.jacc.2019.08.001
Symptoms and Signs of Acute Heart Failure
Manifestations of heart failure differ depending on the extent to which the left ventricle (LV) and right ventricle (RV) are initially affected. Acute RV failure is discussed separately.
History
In LV failure, the most common symptoms are dyspnea and fatigue due to increased pulmonary venous pressures with or without pulmonary edema, and low cardiac output (at rest or inability to augment cardiac output during exertion). Dyspnea may be exertional or occur with rest, depending on the severity of disease. Cardiogenic shock, if present, may cause abnormalities in mental status and organ system function.
Examination
General examination may detect signs of systemic or cardiac disorders that cause or aggravate heart failure (eg, anemia, hyperthyroidism, alcohol use disorder, hemochromatosis, atrial fibrillation with rapid rate, mitral regurgitation).
In LV failure, tachycardia and tachypnea may occur. Heart rate is generally increased as a compensatory mechanism, but bradycardia may occur during decompensation or if there is an underlying bradyarrhythmia. Blood pressure may be high, normal, or low, depending on the changes in cardiac output, volume status, systemic vascular resistance due to the underlying disease, and the compensatory response. Patients with severe LV failure may appear visibly dyspneic or cyanotic, hypotensive, and confused or agitated because of hypoxia and poor cerebral perfusion. Some of these less specific symptoms (eg, confusion) are more common in older patients.
Central cyanosis (affecting all of the body, including warm areas such as the tongue and mucous membranes) reflects severe hypoxemia. Peripheral cyanosis of the lips, fingers, and toes reflects low blood flow with increased oxygen extraction. If vigorous massage improves nail bed color, cyanosis may be peripheral; increasing local blood flow does not improve color if cyanosis is central.
Cardiac findings in acute heart failure with reduced ejection fraction (HFrEF) include:
Diffuse, sustained, and laterally displaced apical impulse
Audible and occasionally palpable third (S3) and fourth (S4) heart sounds
Accentuated pulmonic component (P2) of the second heart sound (S2)
These abnormal heart sounds also can occur in heart failure with preserved ejection fraction (HFpEF) or heart failure with minimally reduced ejection fraction (HRmrEF). A pansystolic murmur of mitral regurgitation at the apex may occur, particularly if the LV is significantly dilated. Jugular venous distention indicates elevated right atrial pressure. Pulsus alternans (an pulse alternating between strong and weak beats) may accompany severely reduced LV systolic function and has historically been considered a poor prognostic sign (1, 2).
Pulmonary findings include early inspiratory basilar crackles that do not clear with coughing and, if pleural effusion is present, dullness to percussion and diminished breath sounds at the lung base(s). Wheezing or stridor may be present due to inflammation of smaller airways as a result of pulmonary edema, or due to compression of larger airways by enlarged cardiac structures (ie, the left atrium or great vessels).
Symptoms and signs references
1. Aslanger E, Aggül B, Albayrak DG. Pulsus Alternans: Caught in Action. Catheter Cardiovasc Interv. 2025;105(1):270-271. doi:10.1002/ccd.31295
2. Schaefer S, Malloy CR, Schmitz JM, Dehmer GJ. Clinical and hemodynamic characteristics of patients with inducible pulsus alternans. Am Heart J. 1988;115(6):1251-1257. doi:10.1016/0002-8703(88)90017-8
Diagnosis of Acute Heart Failure
History and physical examination
Chest radiograph
Echocardiography
Laboratory evaluation, including neurohormonal biomarkers such as brain (B-type) natriuretic peptide (BNP) or N-terminal-pro-BNP (NT-pro-BNP) levels
Sometimes ECG, cardiac radionuclide scan, cardiac MRI, cardiac catheterization, and other tests for etiology and to assess effect on end-organ function
Clinical findings (eg, exertional dyspnea or fatigue, orthopnea, edema, tachycardia, pulmonary crackles, S3, jugular venous distention) suggest heart failure but are usually not apparent early. Some similar symptoms may result from chronic obstructive pulmonary disease (COPD) or recurrent pneumonia or may be erroneously attributed to obesity or old age. Suspicion for heart failure should be high in patients with a history of myocardial infarction, hypertension, valvular disorders, or murmurs and should be moderate in any patient who is older or has diabetes.
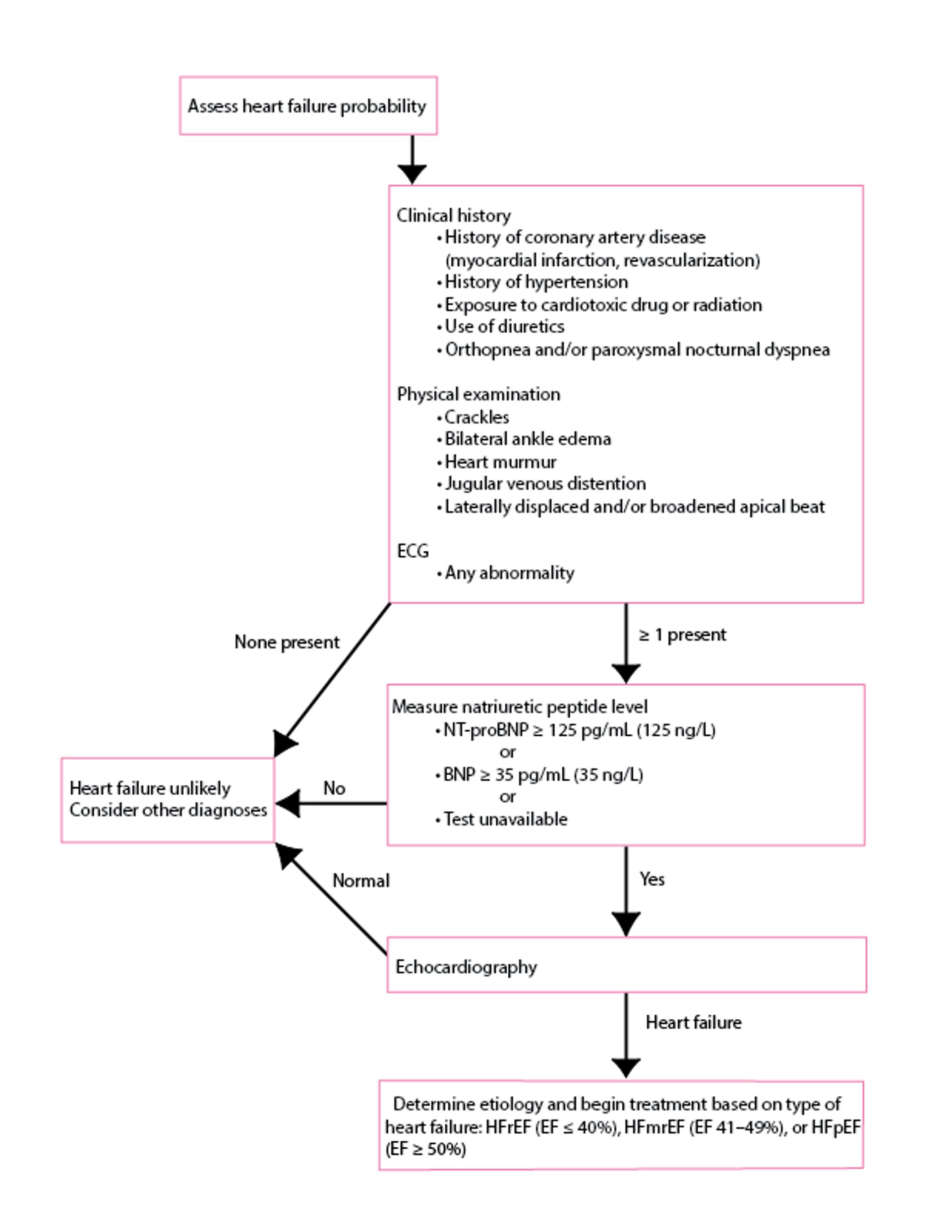
Chest radiograph, ECG, and an objective test of cardiac function, typically echocardiography, should be done (see figure ). Blood tests, except for BNP levels, are not used for diagnosis but are useful for identifying cause and systemic effects (1, 2). Similarly, blood tests, including lactic acid, electrolytes, renal function, and hepatic function, may identify effects on tissue and organ perfusion.
Diagnosis of Acute Heart Failure
Data from McDonagh TA, Metra M, Adamo M, et al: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 42(36):3599-3726, 2021. doi: 10.1093/eurheartj/ehab368. ECG = electrocardiography; EF = ejection fraction; NT-pro-BNP = N-terminal-pro-BNP; BNP = Brain (B-type) natriuretic peptide; HFmrEF = heart failure with mildly reduced ejection fraction; HFpEF = heart failure with preserved ejection fraction; HFrEF = heart failure with reduced ejection fraction. |

Chest radiograph
Chest radiograph findings suggesting heart failure include an enlarged cardiac silhouette, pleural effusion, fluid in the major fissure, and horizontal lines in the periphery of lower posterior lung fields (Kerley B lines). These findings reflect chronic elevation of left atrial pressure and chronic thickening of the intralobular septa due to edema. Upper lobe pulmonary venous congestion and interstitial or alveolar edema may also be present. Careful examination of the cardiac silhouette on a lateral projection can identify specific ventricular and atrial chamber enlargement. The radiograph may also suggest alternative diagnoses (eg, COPD, pneumonia, idiopathic pulmonary fibrosis, lung cancer).
ECG
ECG findings are not diagnostic, but an abnormal ECG, especially one showing previous myocardial infarction, left ventricular hypertrophy, left bundle branch block, or tachyarrhythmia (eg, rapid atrial fibrillation), increases suspicion for heart failure and may help identify the cause. Left or right atrial enlargement may be present. Left ventricular hypertrophy may reflect LV dilation, or voltages may be reduced in the case of severe LV systolic dysfunction.
Other cardiac imaging
Echocardiography can help evaluate chamber dimensions, valve function, LVEF, wall motion abnormalities, LV hypertrophy, diastolic function, pulmonary artery pressure, LV and RV filling pressures, RV function, and pericardial effusion. Intracardiac thrombi, tumors, and calcifications within the heart valves, mitral annulus, and aortic wall abnormalities can be detected. Localized or segmental wall motion abnormalities strongly suggest underlying coronary artery disease but can also be present with patchy myocarditis.
Doppler or color Doppler echocardiography accurately detects valvular disorders and shunts. The combination of Doppler evaluation of mitral inflow with tissue Doppler imaging of the mitral annulus can help identify and quantify LV diastolic dysfunction and LV filling pressures.
Measuring LVEF can distinguish between predominant HFpEF (EF ≥ 50%) and HFrEF (EF ≤ 40%). It is important to re-emphasize that heart failure can occur with a normal LVEF.
Speckle-tracking echocardiography, a measure of strain, is useful in detecting subclinical systolic dysfunction and specific patterns of myocardial dysfunction and may provide useful information regarding the pattern of myocardial involvement of cardiomyopathy, and therefore information about the cause.
Radionuclide imaging also can help assess systolic and diastolic function, previous myocardial infarction, and inducible ischemia or myocardial hibernation. It is used most commonly to assess the presence and/or severity of ischemic heart disease and can also be used to quantify left ventricular ejection fraction.
Cardiac MRI using late gadolinium enhancement imaging (LGE, also called fibrosis or scar imaging) is useful to evaluate the cause of myocardial disease and to detect focal and diffuse myocardial fibrosis. Cardiac amyloidosis, sarcoidosis, hemachromatosis, and myocarditis are causes of HF that can be detected with or suspected based on cardiac MRI findings. Patients with acute heart failure must be stabilized in order to undergo cardiac MRI, because the test requires a prolonged duration in a supine position and coordinated breath holding (or, in some cases, general anesthesia with ventilatory support).
Blood tests
Serum BNP levels are often high in heart failure; this finding may help when clinical findings are unclear or other diagnoses (eg, COPD) need to be excluded. It may be particularly useful for patients with a history of both pulmonary and cardiac disorders. NT-pro-BNP, an inactive moiety created when pro-BNP is cleaved, can be used similarly to BNP. However, a normal BNP level does not exclude the diagnosis of heart failure, particularly in patients with HFpEF and/or obesity. In HFpEF, BNP levels tend to be approximately 50% of those associated with HFrEF (at similar degrees of symptoms), and up to 30% of patients with acute HFpEF have a BNP level below the commonly used threshold of 100 pg/mL (100 ng/L). Obesity, a common comorbidity in HF, is associated with reduced BNP production and increased BNP clearance, resulting in lower levels.
Besides BNP, recommended blood tests include complete blood count, creatinine, blood urea nitrogen (BUN), electrolytes (including magnesium and calcium), glucose, albumin, ferritin, and liver tests. Thyroid function tests are recommended for patients with atrial fibrillation and for selected, especially older, patients.
Other tests
Thoracic ultrasound is a noninvasive method of detecting pulmonary congestion in patients with heart failure. Sonographic "comet tail artifact" on thoracic ultrasound corresponds to the radiographic finding of Kerley B lines.
Coronary angiography or CT coronary angiography is indicated when coronary artery disease is suspected or the etiology of HF is uncertain.
Cardiac catheterization with intracardiac pressure measurements (invasive hemodynamic assessment) may be helpful in the diagnosis of restrictive cardiomyopathies and constrictive pericarditis. Invasive hemodynamic measurements are also very helpful when the diagnosis of HF is equivocal, particularly in patients with HFpEF. In addition, perturbing the cardiovascular system (eg, exercise testing, volume challenge, medication challenges [eg, nitroglycerin, nitroprusside]) can be very helpful during invasive hemodynamic testing to help diagnose HF.with intracardiac pressure measurements (invasive hemodynamic assessment) may be helpful in the diagnosis of restrictive cardiomyopathies and constrictive pericarditis. Invasive hemodynamic measurements are also very helpful when the diagnosis of HF is equivocal, particularly in patients with HFpEF. In addition, perturbing the cardiovascular system (eg, exercise testing, volume challenge, medication challenges [eg, nitroglycerin, nitroprusside]) can be very helpful during invasive hemodynamic testing to help diagnose HF.
Endomyocardial biopsy is sometimes done when an infiltrative cardiomyopathy or acute giant cell myocarditis is strongly suspected but cannot be confirmed with noninvasive imaging (eg, cardiac MRI).
Diagnosis references
1. McDonagh TA, Metra M, Adamo M, et al: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 42(36):3599-3726, 2021. doi: 10.1093/eurheartj/ehab368
2. Heidenreich PA, Bozkurt B, Aguilar D, et al: 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 145:e876–e894, 2022, doi: 10.1161/CIR.0000000000001062
Treatment of Acute Heart Failure
Pharmacotherapy
Respiratory and other organ system support as necessary
Identification and treatment of cause
Sometimes device therapy (eg, implantable cardioverter-defibrillator, cardiac resynchronization therapy, mechanical circulatory support)
Sometimes cardiac transplantation
Multidisciplinary care
Immediate inpatient treatment is required for patients with acute or worsening heart failure (HF) due to certain disorders (eg, acute myocardial infarction, atrial fibrillation with a very rapid ventricular rate, severe hypertension, acute valvular regurgitation), as well as for patients with pulmonary edema, severe symptoms, new-onset HF, or HF unresponsive to outpatient treatment. Patients with mild exacerbations of previously diagnosed HF can be treated at home.
The primary goal is to diagnose and to correct or treat the disorder that led to heart failure.
Acute treatment goals include relieving symptoms and improving hemodynamics (optimizing preload, balancing ventricular function with myocardial oxygen demand, and appropriately reducing afterload); avoiding hypokalemia, renal dysfunction, and symptomatic hypotension; and correcting neurohumoral activation.
The mainstay of the pharmacologic management of acute heart failure is decongestive therapy with diuretics, namely loop diuretics (eg, furosemide, torsemide, bumetanide) in either oral or intravenous form. (See of acute heart failure is decongestive therapy with diuretics, namely loop diuretics (eg, furosemide, torsemide, bumetanide) in either oral or intravenous form. (SeePulmonary Edema for a more detailed discussion.) Loop diuretics may be augmented by adding other diuretic classes such as thiazides (eg, metolazone). Afterload reduction using both oral and intravenous arterial vasodilators (eg, nitroprusside) can also be used to improve acute heart failure. Pulmonary vasodilators (eg, nitrate therapies) may also be used to reduce symptoms of congestion acutely. Low output heart failure, particularly in the setting of cardiogenic shock, often requires acute treatment with intravenous inotropic agents (eg, milrinone, dobutamine). Management of the cause of heart failure may also be indicated acutely, particularly for certain causes such as acute ischemia or arrhythmia.for a more detailed discussion.) Loop diuretics may be augmented by adding other diuretic classes such as thiazides (eg, metolazone). Afterload reduction using both oral and intravenous arterial vasodilators (eg, nitroprusside) can also be used to improve acute heart failure. Pulmonary vasodilators (eg, nitrate therapies) may also be used to reduce symptoms of congestion acutely. Low output heart failure, particularly in the setting of cardiogenic shock, often requires acute treatment with intravenous inotropic agents (eg, milrinone, dobutamine). Management of the cause of heart failure may also be indicated acutely, particularly for certain causes such as acute ischemia or arrhythmia.
For heart failure with reduced ejection fraction (HRrEF) in the acute setting, guideline-directed medical therapy with a sodium-glucose co-transporter 2 (SGLT2) inhibitor, beta-blocker, angiotensin receptor/neprilysin inhibitor, and mineralocorticoid receptor antagonist should be initiated once the patient is stabilized (1, 2). Medication doses are increased rapidly to target doses during the hospitalization and the first 6 weeks of follow-up to reduce the risk of both mortality and re-admission. See Chronic Heart Failure and Medications for Heart Failure for more detailed information on these medications.
Other medications, such as nesiritide (a brain natriuretic peptide), levosimendan (a calcium-sensitizing inotrope), vesnarinone (an inotrope with multiple mechanisms), and ibopamine (a dopaminergic and adrenergic receptor agonist), may provide short-term symptomatic or hemodynamic improvement but do not improve, and in some cases may worsen, long-term outcomes compared to standard therapy (in randomized trials mortality was higher with vesnarinone and ibopamine and similar with nesiritide (3, 4, 5,6). Serelaxin (a recombinant form of the human pregnancy hormone relaxin-2) has been studied, but benefits were not shown in a large international randomized study (7). Omecamtiv mecarbil (an oral cardiac myosin activator) reduced a composite endpoint of heart failure event or death but did not reduce overall cardiovascular mortality in a large randomized trial. (8).
See Medications for Heart Failure for more detailed information on specific medication classes.
Positive airway pressure via high flow nasal cannula, continuous positive airway pressure, or bi-level positive airway pressure, or endotracheal intubation generally reduce left ventricular afterload by increasing intrathoracic pressure and reducing transmural LV pressure.
Arrhythmia treatment
Because arrhythmias can worsen heart failure, it is important to identify and treat the cause of any arrhythmia.
Electrolytes are normalized.
Atrial and ventricular rates are controlled.
Sometimes antiarrhythmic medications are given.
Sinus tachycardia, a common compensatory change in heart failure, usually subsides when HF treatment is effective. If it does not, associated causes (eg, hyperthyroidism, pulmonary emboli, fever, anemia, pain) should be sought. If sinus tachycardia persists despite correction of causes, a beta-blocker, given in gradually increasing doses, may help selected patients. However, lowering heart rate with a beta-blocker can be detrimental to patients with advanced HFpEF (eg, restrictive cardiomyopathy), in whom stroke volume is fixed because of severe diastolic dysfunction. In these patients, cardiac output is heart rate–dependent, and lowering heart rate can thus lower cardiac output at rest with exertion, or both.
Atrial fibrillation with an uncontrolled ventricular rate must be treated; the target resting ventricular rate is typically < 80 beats/minute. Beta-blockers are the treatment of choice, although rate-limiting calcium channel blockers may be used cautiously if systolic function is preserved. Adding digoxin, low-dose amiodarone, or other rhythm and/or rate controlling medications may help some patients. Routine conversion to and maintenance of sinus rhythm has not been shown to be superior to rate control alone in randomized trials (80 beats/minute. Beta-blockers are the treatment of choice, although rate-limiting calcium channel blockers may be used cautiously if systolic function is preserved. Adding digoxin, low-dose amiodarone, or other rhythm and/or rate controlling medications may help some patients. Routine conversion to and maintenance of sinus rhythm has not been shown to be superior to rate control alone in randomized trials (9). However, it is best to make this determination on a case-by-case basis because some patients improve significantly with restoration of normal sinus rhythm. If rapid atrial fibrillation does not respond to medication, permanent pacemaker insertion with complete or partial ablation of the atrioventricular node, or other atrial fibrillation ablation procedures, may be considered to restore a sinus or regular rhythm in selected patients.
Isolated ventricular premature beats, which are common in HF, do not require specific treatment, although rarely very frequent ventricular premature beats (> 15,000/day) have been shown to precipitate heart failure (that reverses with suppression). However, optimization of HF treatments and correction of electrolyte abnormalities (especially of potassium and magnesium) reduce the risk of ventricular arrhythmias.
Sustained ventricular tachycardia that persists despite correction of cause (eg, low potassium or magnesium, ischemia) and optimal medical treatment of HF may require an antiarrhythmic medication. Amiodarone, beta-blockers, and dofetilide are the medications of choice because other antiarrhythmics have adverse proarrhythmic effects when LV systolic dysfunction is present. that persists despite correction of cause (eg, low potassium or magnesium, ischemia) and optimal medical treatment of HF may require an antiarrhythmic medication. Amiodarone, beta-blockers, and dofetilide are the medications of choice because other antiarrhythmics have adverse proarrhythmic effects when LV systolic dysfunction is present.
Because amiodarone increases digoxin and warfarin levels, increases digoxin and warfarin levels,digoxin and warfarin doses should be decreased by half or stopped. Serum digoxin level and INR (international normalized ratio) level should be routinely monitored. However, medication toxicity can occur even at therapeutic levels. Because long-term use of amiodarone can cause adverse effects, a low dose is used when possible; blood tests for liver function and thyroid-stimulating hormone are done every 6 months. If the chest radiograph is abnormal or dyspnea worsens significantly, chest radiographs and pulmonary function tests are done yearly to check for pulmonary fibrosis. For sustained ventricular arrhythmias, amiodarone may be required; to reduce risk of sudden death, an oral loading dose is given twice a day for 1 to 3 weeks until rhythm control is adequate, then dose is decreased over 1 month to a once daily maintenance dose.
Device therapy and mechanical circulatory support
Device therapy, including implantable cardioverter-defibrillator (ICD) and cardiac resynchronization therapy (CRT), is primarily used for chronic heart failure. In the acute setting, however, CRT may provide symptom management, and both CRT and ICD may be used in the management of a refractory arrhythmia causing or exacerbating the heart failure.
Ultrafiltration (venovenous filtration) can be useful in selected hospitalized patients with severe cardiorenal syndrome and volume overload refractory to diuretics. However, ultrafiltration should not be used routinely because evidence does not clearly show long-term clinical benefit (10).
An intra-aortic counterpulsation balloon pump (IABP) is helpful in selected patients with acute HF who have a good chance of recovery (eg, acute HF following myocardial infarction) or in those who need a bridge to a more permanent solution such as cardiac surgery (eg, to fix severe valvular disease or to revascularize multivessel coronary artery disease), an LV assist device, or heart transplantation.
Other forms of temporary mechanical circulatory support for patients with acute HF and cardiogenic shock include surgically placed devices such as extracorporeal membrane oxygenation (ECMO, typically with venoarterial cannulation in patient's with heart failure) and centrifugal flow ventricular assist devices that can support either the LV, the RV, or both and can also be combined with an oxygenator to provide full cardiopulmonary support. Percutaneously placed devices such as intravascular microaxial ventricular assist devices are available for both LV and RV support. Selection of temporary mechanical circulatory support devices is based mainly on availability and local medical center experience.
Please see device therapy for chronic heart failure for more information.
Prognosis for Acute Heart Failure
Generally, patients hospitalized for acute heart failure have a poor prognosis unless the cause is correctable. Overall combined 5-year survival is estimated at 34%, and 10-year survival at 17%, for patients with after an initial hospitalization for all types of heart failure (1).
Prognosis reference
1. Hariharaputhiran S, Peng Y, Ngo L, et al. Long-term survival and life expectancy following an acute heart failure hospitalization in Australia and New Zealand. Eur J Heart Fail. 2022;24(9):1519-1528. doi:10.1002/ejhf.2595
Key Points
Consider acute heart failure in patients with worsening or new onset of exertional dyspnea or fatigue, orthopnea, and/or edema, particularly in those with a history of myocardial infarction, hypertension, or valvular disorders or murmurs.
Perform chest radiograph, ECG, brain (B-type) natriuretic peptide levels, echocardiography, and additional testing as necessary.
Treat with guideline-directed medical therapy appropriate to heart failure type prior to hospital discharge.
Treatment includes control of underlying disorders, medications, sometimes implantable devices (cardiac resynchronization therapy, implantable cardioverter-defibrillators), education and lifestyle changes.
Drug Information for the Topic





