

Shock is a state of organ hypoperfusion with resultant cellular dysfunction and damage. Mechanisms may involve decreased circulating volume, decreased cardiac output, and vasodilation, sometimes with shunting of blood to bypass capillary exchange beds. Symptoms include altered mental status, tachycardia, hypotension, and oliguria. Diagnosis is mostly clinical, based on a characteristic combination of signs and symptoms (including hypotension, tachycardia, tachypnea, oliguria, and obtundation) and sometimes supported by measurement of markers of tissue hypoperfusion (eg, blood lactate, base deficit). Treatment is with fluid resuscitation, including blood products if necessary, correction of the underlying disorder, and sometimes vasopressors.
(See also Sepsis and Septic Shock.)
Pathophysiology of Shock
The fundamental defect in shock is reduced perfusion of vital tissues. Once perfusion declines and oxygen delivery to cells is inadequate for aerobic metabolism, cells shift to anaerobic metabolism with increased production of carbon dioxide and elevated blood lactate levels. Cellular function declines, and if shock persists, irreversible cell damage and death occur.
During shock, both the inflammatory and clotting cascades may be triggered in areas of hypoperfusion. Hypoxic vascular endothelial cells activate white blood cells, which bind to the endothelium and release directly damaging substances (eg, reactive oxygen species, proteolytic enzymes) and inflammatory mediators (eg, cytokines, leukotrienes, tumor necrosis factor). Some of these mediators bind to cell surface receptors and activate nuclear factor kappa B (NFκB), which leads to production of additional cytokines and nitric oxide (NO), a potent vasodilator. B), which leads to production of additional cytokines and nitric oxide (NO), a potent vasodilator.Disseminated intravascular coagulation can sometimes result from activation of these cascades.
There are various mechanisms of shock, described below. Septic shock, a type of distributive shock, may be more proinflammatory than other forms of shock because of the actions of bacterial toxins, especially endotoxin.
Blood pressure is not always low in the early stages of shock (although hypotension eventually occurs if shock is not reversed). Similarly, not all patients with “low” blood pressure have shock. The degree and consequences of hypotension vary with the adequacy of physiologic compensation and the patient’s underlying diseases. Thus, a modest degree of hypotension that is well tolerated by a younger or relatively healthy person might result in severe cerebral, cardiac, or renal dysfunction in an older adult with significant arteriosclerosis.
Compensation for shock
Initially, when oxygen delivery (DO2) is decreased, tissues compensate by extracting a greater percentage of delivered oxygen. Low arterial pressure triggers an adrenergic response with sympathetic-mediated vasoconstriction and often increased heart rate. Initially, vasoconstriction is selective, shunting blood to the heart and brain and away from the splanchnic circulation. Circulating beta-adrenergic amines (epinephrine, norepinephrine) also increase cardiac contractility and trigger release of
Corticosteroids from the adrenal gland
Renin from the kidneys
Glucose from the liver
Corticosteroids enhance the effects of catecholamines. Renin stimulates volume retention and vasoconstriction. Increased glucose increases pyruvate uptake in the mitochondria, which increases lactate production when there is insufficient oxygen.
Reperfusion following shock
With resolution of shock, reperfusion of ischemic cells can cause further injury. As substrate is reintroduced, neutrophil activity may increase, increasing production of damaging superoxide and hydroxyl radicals. After blood flow is restored, locally concentrated inflammatory mediators may be circulated to other organs.
Multiple organ dysfunction syndrome (MODS)
The combination of direct and reperfusion injury may cause MODS—the progressive dysfunction of ≥ 2 organs due to life-threatening illness or injury. MODS can follow any type of shock but is most common when infection is involved; organ failure is one of the defining features of septic shock. MODS also occurs in > 10% of patients with severe traumatic injury and is the primary cause of death in those surviving > 24 hours.
Pearls & Pitfalls
|
Any organ system can be affected, but the most frequent target organs are the lungs, in which increased membrane permeability leads to flooding of alveoli and further inflammation. Progressive hypoxia may be increasingly resistant to supplemental oxygen therapy. This condition is termed acute lung injury or, if severe, acute respiratory distress syndrome (ARDS).
The kidneys are injured when renal perfusion is critically reduced, leading to acute tubular necrosis and renal insufficiency manifested by oliguria and a progressive rise in serum creatinine.
In the heart, reduced coronary perfusion and increased inflammatory mediators (including tumor necrosis factor and interleukin-1) may depress contractility, worsen myocardial compliance, and down-regulate beta-receptors. Arrhythmias may occur. These factors decrease cardiac output, further worsening both myocardial and systemic perfusion and causing a vicious circle that often culminates in death.
In the gastrointestinal tract, ileus and submucosal hemorrhage can develop. Liver hypoperfusion can cause focal or extensive hepatocellular necrosis, transaminase and bilirubin elevation, and decreased production of clotting factors.
Coagulation can be impaired, including the most severe manifestation, disseminated intravascular coagulopathy.
Etiology and Classification of Shock
There are several mechanisms of organ hypoperfusion and shock. Shock may be due to
A low circulating volume (hypovolemic shock)
Vasodilation (distributive shock)
A primary decrease in cardiac output (both cardiogenic and obstructive shock)
A combination

Hypovolemic shock
Hypovolemic shock is caused by a critical decrease in intravascular volume. Diminished venous return (preload) results in decreased ventricular filling and reduced stroke volume. Unless compensated for by increased heart rate, cardiac output decreases.
A common cause is bleeding (hemorrhagic shock), typically due to trauma, surgical interventions, peptic ulcer, esophageal varices, or ruptured aortic aneurysm. Bleeding may be overt (eg, hematemesis, melena) or concealed (eg, ruptured ectopic pregnancy).
Hypovolemic shock may also follow increased losses of body fluids other than blood (nonhemorrhagic, see table Hypovolemic Shock Caused by Body Fluid Loss).
Hypovolemic Shock Caused by Body Fluid Loss (Nonhemorrhagic)
Site of Fluid Loss | Mechanism of Loss |
|---|---|
Skin | Thermal or chemical burn Sweating due to excessive heat exposure |
Gastrointestinal tract | |
Kidneys | Diabetes mellitus, arginine vasopressin deficiency (formerly central diabetes insipidus), or arginine vasopressin resistance (formerly nephrogenic diabetes insipidus) Polyuric phase after acute tubular damage Potent diuretic use Salt-losing nephritis |
Intravascular fluid lost to the extravascular space | Increased capillary permeability secondary to inflammation, severe systemic hypoxia or ischemia, or traumatic injury (eg, crush), sepsis, bowel ischemia, acute pancreatitis |
Hypovolemic shock may be due to inadequate fluid intake (with or without increased fluid loss). Water may be unavailable, neurologic disability may impair the thirst mechanism, or physical disability may impair access.
In hospitalized patients, hypovolemia can be compounded if early signs of circulatory insufficiency are incorrectly ascribed to heart failure and fluids are withheld or diuretics are given.
Distributive shock
Distributive shock results from a relative inadequacy of intravascular volume caused by arterial or venous vasodilation; circulating blood volume is normal. In some cases, cardiac output (and DO2) is high, but increased blood flow through arteriovenous shunts bypasses capillary beds; this bypass plus uncoupled cellular oxygen transport cause cellular hypoperfusion (shown by decreased oxygen consumption). In other situations, blood pools in venous capacitance beds and cardiac output falls.
Distributive shock may be caused by
Anaphylaxis (anaphylactic shock)
Bacterial infection with endotoxin release (septic shock) or exotoxin release (toxic shock)
Severe injury to the spinal cord, usually above T4 (neurogenic shock)
Ingestion of certain medications (eg, nitrates, opioids, or adrenergic blockers) or poisons (toxin-induced shock)
In septic shock, vasodilation of capacitance vessels leads to pooling of blood and hypotension because of “relative” hypovolemia (ie, too much volume to be filled by the existing amount of blood). Localized vasodilation may shunt blood past the capillary exchange beds, causing focal hypoperfusion despite normal cardiac output and blood pressure. Additionally, excess nitric oxide is converted to peroxynitrite, a free radical that damages mitochondria and decreases ATP (In septic shock, vasodilation of capacitance vessels leads to pooling of blood and hypotension because of “relative” hypovolemia (ie, too much volume to be filled by the existing amount of blood). Localized vasodilation may shunt blood past the capillary exchange beds, causing focal hypoperfusion despite normal cardiac output and blood pressure. Additionally, excess nitric oxide is converted to peroxynitrite, a free radical that damages mitochondria and decreases ATP (adenosine triphosphate) production. Blood flow to microvessels, including capillaries, is reduced even though large-vessel blood flow is preserved in septic shock. Mechanical microvascular obstruction may, at least in part, account for such limiting of substrate delivery. Leukocytes and platelets adhere to the endothelium as part of the clotting cascade, causing the conversion of fibrinogen to fibrin and stabilizing the platelet plug.
Multiple mediators, along with endothelial cell dysfunction, markedly increase microvascular permeability, allowing fluid and sometimes plasma proteins to escape into the interstitial space (1, 2, 3). In the gastrointestinal tract, increased permeability possibly allows translocation of the enteric bacteria from the lumen, potentially leading to sepsis or metastatic infection.
Neutrophil apoptosis may be inhibited, enhancing the release of inflammatory mediators. In other cells, apoptosis may be augmented, increasing cell death and thus worsening organ function.
Anaphylactic shock and septic shock often have a component of hypovolemia as well.
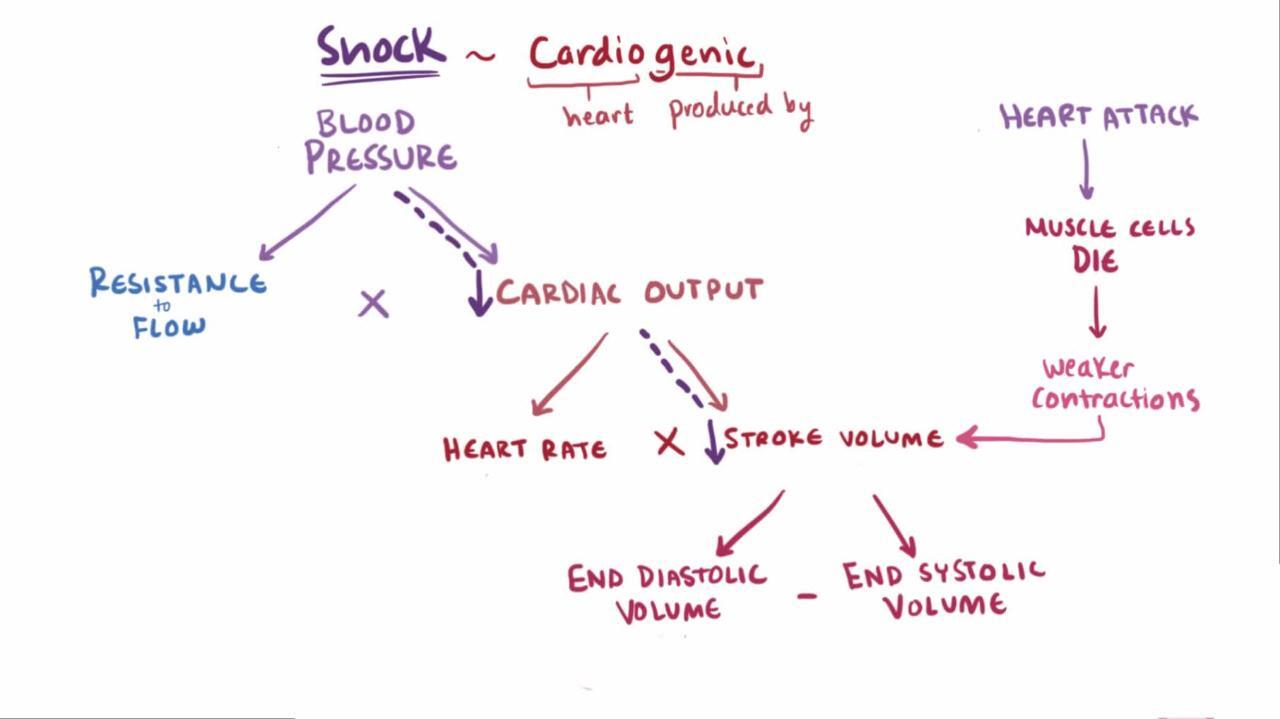
Cardiogenic and obstructive shock
Cardiogenic shock is a relative or absolute reduction in cardiac output due to a primary cardiac disorder. Obstructive shock is caused by mechanical factors that interfere with filling or emptying of the heart or great vessels. Causes are listed in the table Mechanisms of Cardiogenic and Obstructive Shock.
Mechanisms of Cardiogenic and Obstructive Shock
Type | Mechanism | Cause |
|---|---|---|
Obstructive | Mechanical interference with ventricular filling | Tension pneumothorax, vena cava compression, cardiac tamponade, atrial tumor or clot |
Interference with ventricular emptying | ||
Cardiogenic | Impaired myocardial contractility | Myocardial ischemia or myocardial infarction, myocarditis, medications |
Abnormalities of cardiac rhythm | Tachycardia, bradycardia | |
Cardiac structural disorder | Acute mitral or aortic regurgitation, ruptured interventricular septum, prosthetic valve malfunction |
Etiology and classification references
1. Salmon AH, Satchell SC: Endothelial glycocalyx dysfunction in disease: Albuminuria and increased microvascular permeability. J Pathol 226:562–74, 2012. doi: 10.1002/path.3964
2. Chelazzi C, Villa G, Mancinelli P, et al: Glycocalyx and sepsis-induced alterations in vascular permeability. Crit Care 19(1):26, 2015. doi:10.1186/s13054-015-0741-z
3. Martin L, Koczera P, Zechendorf E, et al: The endothelial glycocalyx: New diagnostic and therapeutic approaches in sepsis. Biomed Res Int 2016:3758278, 2016. doi:10.1155/2016/3758278
Symptoms and Signs of Shock
Altered mental status (eg, lethargy, confusion, somnolence) is a common sign of shock. The hands and feet are pale, cool, clammy, and often cyanotic, as are the earlobes, nose, and nail beds. Capillary filling time is prolonged, and, except in distributive shock, the skin appears grayish or dusky and moist. Overt diaphoresis may occur. Peripheral pulses are weak and typically rapid; often, only femoral or carotid pulses are palpable. Tachypnea and hyperventilation may be present. Blood pressure tends to be low (< 90 mm Hg systolic) or unobtainable; direct measurement by intra-arterial catheter, if done, often gives higher and more accurate values. Urine output is low.
Distributive shock causes similar symptoms, except the skin may appear warm or flushed, especially during sepsis. The pulse may be bounding rather than weak. In septic shock, fever, possibly preceded by chills, is typically present. Some patients with anaphylactic shock have urticaria or wheezing.
Numerous other symptoms (eg, chest pain, dyspnea, abdominal pain) may be due to the underlying disease or secondary organ failure.
Diagnosis of Shock
History and physical examination
Vital signs
Test result trends
Diagnosis is mostly clinical, based on evidence of insufficient tissue perfusion (depressed levels of consciousness, oliguria, peripheral cyanosis) and signs of compensatory mechanisms (tachycardia, tachypnea, diaphoresis). Specific criteria include
Obtundation
Heart rate > 100 beats/minute
Respiratory rate > 22 breaths/minute
Hypotension (systolic blood pressure < 90 mm Hg) or a 30-mm Hg fall in baseline blood pressure
Urine output < 0.5 mL/kg/hour
Laboratory findings that support the diagnosis include
Lactate > 3 mmol/L (27 mg/dL)
Base deficit < −4 mEq/L
PaCO2 < 32 mm Hg (< 4.26 kPa)
However, none of these findings alone is diagnostic, and each is evaluated by its trend (ie, worsening or improving) and in the overall clinical context, including physical signs.
Near-infrared spectroscopy is a noninvasive and rapid technique that may measure the degree of shock; however, this technique has yet to be validated on a larger scale.
Identifying etiology
Recognizing the cause of shock is often more important than categorizing the type. The cause may be obvious or can be recognized quickly based on the history and physical examination, aided by simple testing.
Causes may be related to the heart or lungs:
Chest pain (with or without dyspnea) suggests myocardial infarction (MI), aortic dissection, or pulmonary embolism.
A systolic murmur may indicate ventricular septal rupture or mitral regurgitation due to acute MI.
A diastolic murmur may indicate aortic regurgitation due to aortic dissection involving the aortic root.
Cardiac tamponade is suggested by jugular venous distention, muffled heart sounds, and a paradoxical pulse.
Pulmonary embolism severe enough to cause shock typically produces decreased oxygen saturation and occurs more often in special settings, including prolonged bed rest and after a surgical procedure.
Tests to evaluate for and differentiate between these cardiac or pulmonary causes include electrocardiography (ECG), cardiac enzyme measurement, chest x-ray, arterial blood gas (ABG) measurement, lung scan, helical CT, and echocardiography.
Causes may be related to problems in the abdomen:
Abdominal or back pain or a tender abdomen suggests pancreatitis, ruptured abdominal aortic aneurysm, peritonitis (eg, due to a perforated viscus), and, in women of childbearing age, ruptured ectopic pregnancy.
A pulsatile midline mass suggests ruptured abdominal aortic aneurysm. A tender adnexal mass suggests ectopic pregnancy.
Tests typically include abdominal CT (if the patient is unstable, bedside ultrasound can be helpful), complete blood count (CBC), amylase, lipase, and, for women of childbearing age, urine pregnancy test.
Other causes include
Fever, chills, and focal signs of infection suggest septic shock, particularly in patients with immunosuppression.
Isolated fever, contingent on history and clinical settings, may point to heatstroke.
Tests include chest x-ray; urinalysis; CBC; and cultures of wounds, blood, urine, and other relevant body fluids.
In a few patients, the cause is occult. Patients with no focal symptoms or signs indicative of cause should have ECG, cardiac enzyme measurement, chest x-ray, and ABGs. If results of these tests are normal, other potential causes include medication or illicit drug overdose, occult infection (including toxic shock), anaphylaxis, and obstructive shock.
Additional testing
If not already done, ECG, chest x-ray, CBC, serum electrolytes, blood urea nitrogen (BUN), creatinine, prothrombin time (PT), partial thromboplastin time (PTT), liver function tests, and fibrinogen and fibrin split products are done to monitor the patient's status and serve as a baseline.
If the patient’s volume status is difficult to determine, monitoring of central venous pressure (CVP) or pulmonary artery occlusion pressure (PAOP) may be useful. CVP < 5 mm Hg (< 7 cm water) or PAOP < 8 mm Hg may indicate hypovolemia, although CVP may be greater in patients with hypovolemia who have preexisting pulmonary hypertension.
Rapid bedside cardiac ultrasound (done by the treating physician) to assess adequacy of cardiac filling and function is often used to assess shock and overall cardiac performance (for review, 1, 2).
Diagnosis references
1. Ferrada P: Image-based resuscitation of the hypotensive patient with cardiac ultrasound: an evidence-based review. J Trauma Acute Care Surg 80 (3): 511–518, 2016.
2. Martin ND, Codner P, Greene W, et al: Contemporary hemodynamic monitoring, fluid responsiveness, volume optimization, and endpoints of resuscitation: an AAST critical care committee clinical consensus. Trauma Surg Acute Care Open 5(1):e000411, 2020. doi: 10.1136/tsaco-2019-000411
Treatment of Shock
Supportive care
IV fluids
Other management depending on type and cause of shock
General management of shock
First aid involves keeping the patient warm. External hemorrhage is controlled, airway and ventilation are checked, and respiratory assistance is given if necessary. Nothing is given by mouth, and the patient’s head is turned to one side to avoid aspiration if emesis occurs.
Treatment begins simultaneously with evaluation.
Supplemental oxygen by face mask is provided. If shock is severe or if ventilation is inadequate, airway intubation with mechanical ventilation is necessary. Two large (14- to 16-gauge) IV catheters are inserted into separate peripheral veins. A central venous line or an intraosseous needle, especially in children, provides an alternative when peripheral veins cannot promptly be accessed.
Typically, a fluid challenge is given; 1 L (or 20 mL/kg in children) of 0.9% saline is infused over 15 minutes. In major hemorrhage, Ringer’s lactate is commonly used, although in major hemorrhage, use of crystalloid should be minimized in favor of transfusion of blood products (red blood cells, fresh frozen plasma, and platelets in a 1:1:1 ratio) (1, 2). Unless clinical parameters return to normal, the infusion of fluid is continued. Smaller volumes (eg, 250 to 500 mL) are used for patients with signs of high right-sided pressure (eg, distention of neck veins) or acute myocardial infarction.
A fluid challenge should probably not be done in a patient with signs of pulmonary edema. Further fluid therapy is based on the underlying condition and may require monitoring of CVP or PAOP. Bedside cardiac ultrasound to assess contractility and vena caval respiratory variability may help determine the need for additional fluid vs the need for inotropic support.
Patients in shock are critically ill and should be admitted to an intensive care unit. Monitoring includes
ECG
Systolic, diastolic, and mean blood pressure, preferably by intra-arterial catheter
Respiratory rate and depth
Pulse oximetry
Urine flow by indwelling bladder catheter
Body temperature
Clinical status, including sensorium (eg, Glasgow Coma Scale), pulse volume, skin temperature, and color
Measurement of CVP, PAOP, and thermodilution cardiac output using a balloon-tipped pulmonary arterial catheter may be helpful for diagnosis and initial management of patients with shock of uncertain or mixed etiology or with severe shock, especially when accompanied by oliguria or pulmonary edema. Echocardiography (bedside or transesophageal) is a less invasive alternative.
Serial measurements of arterial blood gases, hematocrit, electrolytes, serum creatinine, and blood lactate are obtained. Sublingual carbon dioxide measurement, if available, is a noninvasive monitor of visceral perfusion (levels increase with decreasing tissue perfusion). A well-designed flow sheet to monitor trends is helpful.
Because tissue hypoperfusion makes intramuscular absorption unreliable, all parenteral medications are given IV. Opioids generally are avoided because they may cause vasodilation, but severe pain may be treated with morphine 0.1 mg/kg IV given over 2 minutes and repeated every 10 to 15 minutes if necessary. Although cerebral hypoperfusion may cause anxiety, sedatives or tranquilizers are not routinely given unless the patient is intubated.Because tissue hypoperfusion makes intramuscular absorption unreliable, all parenteral medications are given IV. Opioids generally are avoided because they may cause vasodilation, but severe pain may be treated with morphine 0.1 mg/kg IV given over 2 minutes and repeated every 10 to 15 minutes if necessary. Although cerebral hypoperfusion may cause anxiety, sedatives or tranquilizers are not routinely given unless the patient is intubated.
After initial resuscitation, specific treatment is directed at the underlying condition. Additional supportive care is guided by the type of shock.

Treatment of hemorrhagic shock
Surgical control of bleeding
Early transfusion of blood products
In hemorrhagic shock, surgical control of bleeding is the first priority. Volume replacement accompanies rather than precedes surgery to restore hemostasis. Blood products and crystalloid solutions are used for resuscitation; however, red blood cells, fresh frozen plasma, and platelets are being given earlier and in a ratio of 1:1:1 in patients likely to require massive transfusion. Failure to respond usually indicates insufficient volume administration or unrecognized ongoing hemorrhage. Vasopressors may be tried in refractory hemorrhagic shock but only after adequate blood volume has been restored and hemorrhage controlled; giving vasopressors before that can worsen outcomes.
Treatment of distributive shock
IV crystalloids
Sometimes inotropic or vasopressor drugs
Antimicrobials for septic shock
Epinephrine for anaphylaxisEpinephrine for anaphylaxis
Distributive shock with profound hypotension after initial fluid replacement with 0.9% saline may be treated with inotropic or vasopressor agents (eg, dopamine, norepinephrine—see table Distributive shock with profound hypotension after initial fluid replacement with 0.9% saline may be treated with inotropic or vasopressor agents (eg, dopamine, norepinephrine—see tableInotropic and Vasoactive Catecholamines). Patients with septic shock also receive broad-spectrum antibiotics. Patients with anaphylactic shock unresponsive to fluid challenge (especially if accompanied by bronchoconstriction) receive epinephrine 0.05 to 0.1 mg IV, followed by unresponsive to fluid challenge (especially if accompanied by bronchoconstriction) receive epinephrine 0.05 to 0.1 mg IV, followed byepinephrine infusion 0.02 mcg/kg/minute.
Inotropic and Vasoactive Catecholamines
Medication | Dosage | Hemodynamic Actions |
|---|---|---|
DobutamineDobutamine | 2.5–10 mcg/kg/minute | Beta-adrenergic: Inotropic effects* |
DopamineDopamine | 2–10 mcg/kg/minute for low dose 20 mcg/kg/minute for high dose | Alpha-adrenergic: Vasoconstriction† Beta-adrenergic: Inotropic and chronotropic effects and vasodilation† Nonadrenergic: Renal and splanchnic vasodilation |
NorepinephrineNorepinephrine | 2–12 mcg/minute OR 0.1 mcg/kg/minute (weight-based dosing) | Alpha-adrenergic: Vasoconstriction Beta-adrenergic: Inotropic and chronotropic effects |
* Chronotropic, arrhythmogenic, and direct vascular effects are minimal at lower doses. | ||
† Effects depend on dosage and underlying pathophysiology. | ||
Treatment of cardiogenic shock
Treatment of cause
In cardiogenic shock, structural disorders (eg, valvular dysfunction, septal rupture) are repaired surgically.
Coronary thrombosis is treated either by percutaneous interventions (angioplasty, stent placement), coronary artery bypass surgery, or thrombolysis.
Tachydysrhythmia (eg, rapid atrial fibrillation, ventricular tachycardia) is slowed by cardioversion or with antiarrhythmic medications. Bradycardia is treated with a transcutaneous or transvenous pacemaker; atropine 0.5 mg IV every 5 minutes up to 4 doses may be given pending pacemaker placement. Isoproterenol at 1 to 4 mcg/minute is occasionally useful if atropine is ineffective, but it is not advised in patients with myocardial ischemia due to coronary artery disease.; atropine 0.5 mg IV every 5 minutes up to 4 doses may be given pending pacemaker placement. Isoproterenol at 1 to 4 mcg/minute is occasionally useful if atropine is ineffective, but it is not advised in patients with myocardial ischemia due to coronary artery disease.
Shock after acute MI is treated with volume expansion if PAOP is low or normal; 15 to 18 mm Hg is considered optimal. If a pulmonary artery catheter is not in place or bedside cardiac ultrasound is not available, cautious volume infusion (250- to 500-mL bolus of 0.9% saline) may be tried while auscultating the chest frequently for signs of fluid overload. Shock after right ventricular MI usually responds partially to volume expansion; however, vasopressor agents may be needed. Bedside cardiac ultrasound to assess contractility and vena caval respiratory variability can help determine the need for additional fluid or vasopressors; inotropic support is a better approach for patients with normal or above-normal filling.
If hypotension is moderate (eg, mean arterial pressure [MAP] 70 to 90 mm Hg), dobutamine infusion may be used to improve cardiac output and reduce left ventricular filling pressure. Tachycardia and arrhythmias occasionally occur during If hypotension is moderate (eg, mean arterial pressure [MAP] 70 to 90 mm Hg), dobutamine infusion may be used to improve cardiac output and reduce left ventricular filling pressure. Tachycardia and arrhythmias occasionally occur duringdobutamine administration, particularly at higher doses, necessitating dose reduction. Vasodilators (eg, nitroprusside, nitroglycerin), which increase venous capacitance or lower systemic vascular resistance, reduce the workload on the damaged myocardium and may increase cardiac output in patients without severe hypotension. Combination therapy (eg, dopamine or administration, particularly at higher doses, necessitating dose reduction. Vasodilators (eg, nitroprusside, nitroglycerin), which increase venous capacitance or lower systemic vascular resistance, reduce the workload on the damaged myocardium and may increase cardiac output in patients without severe hypotension. Combination therapy (eg, dopamine ordobutamine with nitroprusside or nitroglycerin) may be particularly useful but requires close ECG and pulmonary and systemic hemodynamic monitoring.
For more serious hypotension (MAP < 70 mm Hg), norepinephrine or dopamine may be given, with a target systolic pressure of 80 to 90 mm Hg (and not 70 mm Hg), norepinephrine or dopamine may be given, with a target systolic pressure of 80 to 90 mm Hg (and not> 110 mm Hg).
Intra-aortic balloon counterpulsation is valuable for temporarily reversing shock in patients with acute MI. This procedure should be considered as a bridge to permit cardiac catheterization and coronary angiography before possible surgical intervention in patients with acute MI complicated by ventricular septal rupture or severe acute mitral regurgitation who require vasopressor support for > 30 minutes.
In obstructive shock, nontraumatic cardiac tamponade requires immediate pericardiocentesis, which can be done at the bedside. Trauma-related cardiac tamponade requires surgical decompression and repair.
Tension pneumothorax should be immediately decompressed with a catheter inserted into the 2nd intercostal space, midclavicular line; a chest tube is then inserted.
Massive pulmonary embolism resulting in shock is treated with anticoagulation and thrombolysis, surgical embolectomy, or extracorporeal membrane oxygenation in select cases.
Treatment references
1. Holcomb JB, Tilley BC, Baraniuk S, et al: Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: The PROPPR randomized clinical trial. JAMA 313(5):471-482, 2015. doi:10.1001/jama.2015.12
2. Cannon JW, Khan MA, Raja AS, et al: Damage control resuscitation in patients with severe traumatic hemorrhage: A practice management guideline from the Eastern Association for the Surgery of Trauma. J Trauma Acute Care Surg 82(3): 605-617, 2017. doi: 10.1097/TA.0000000000001333
Prognosis for Shock
Untreated shock is usually fatal. Even with treatment, mortality from cardiogenic shock after MI (60 to 65%) and septic shock (30 to 40%) is high.
Prognosis depends on the cause, preexisting or complicating illness, time between onset and diagnosis, and promptness and adequacy of therapy.
Drugs Mentioned In This Article





