

Dyslipidemia is elevation of plasma cholesterol, triglycerides (TGs), or both, or a low high-density lipoprotein cholesterol (HDL-C) level that contributes to the development of atherosclerosis. Causes may be primary (genetic) or secondary. Diagnosis is by measuring plasma levels of total cholesterol, triglycerides, and individual lipoproteins. Treatment involves dietary changes, exercise, and lipid-lowering medications.
(See also Overview of Lipid Metabolism.)
Serum lipid levels are continuous; there is no precise threshold between normal and abnormal levels. A linear relation likely exists between lipid levels and cardiovascular risk (see table ), so many people with “normal” cholesterol levels benefit from achieving still lower levels (1). Consequently, there are no numeric definitions of dyslipidemia; the term is applied to lipid levels for which treatment has proven beneficial.
Proof of benefit is strongest for lowering elevated low-density lipoprotein cholesterol (LDL-C) levels. In the general population, evidence is less strong for a benefit from lowering elevated triglyceride and increasing low high-density lipoprotein cholesterol (HDL-C) levels.
HDL-C levels do not always predict cardiovascular risk. For example, high HDL-C levels caused by some genetic disorders may not be associated with a lower risk of cardiovascular disorders, and low HDL-C levels caused by some genetic disorders may not be associated with an increased risk of cardiovascular disorders. Although low HDL-C levels predict cardiovascular risk in the general population, the increased risk may be caused by other factors, such as accompanying lipid and metabolic abnormalities like hypertriglyceridemia, rather than the HDL-C level itself (2-8). Furthermore, some evidence supports a U-shaped relationship between HDL-C levels and adverse cardiovascular outcomes, with elevated risk at both the lowest and highest HDL-C concentrations (9).
Cholesterol Levels and Cardiovascular Risk
Cardiovascular Risk | Total Cholesterol | LDL-C | HDL-C |
|---|---|---|---|
Higher risk | ≥ 6.2 mmol/L (240 mg/dL) | ≥ 4.1 mmol/L (160 mg/dL) | Male: < 1.0 mmol/L (40 mg/dL) Female: < 1.3 mmol/L (50 mg/dL) |
At- risk | 5.2-6.2 mmol/L (200-239 mg/dL) | 2.6-4.1 mmol/L (100-159 mg/dL) | Male: 1.0-1.5 mmol/L (40-59 mg/dL) Female: 1.3-1.5 mmol/L (50-59 mg/dL) |
Lower risk | < 5.2 mmol/L (200 mg/dL) | < 2.6 mmol/L (100 mg/dL) | ≥ 1.6 mmol/L (60 mg/dL )* |
* Evidence supports a U-shaped curve between HDL levels and cardiac risk, with possible increased risk associated with very elevated HDL. (Mørland JG, Magnus P, Vollset SE, Leon DA, Selmer R, Tverdal A. Associations between serum high-density lipoprotein cholesterol levels and cause-specific mortality in a general population of 345 000 men and women aged 20-79 years. Int J Epidemiol 2023;52(4):1257-1267. doi:10.1093/ije/dyad011) | |||
HDL = high-density lipoprotein; LDL = low-density lipoprotein. | |||
Data from Carmena R: Primary Mixed Dyslipidemias, Editor(s): Ilpo Huhtaniemi, Luciano Martini, Encyclopedia of Endocrine Diseases (Second Edition), Academic Press, 2019, Pages 314-319, ISBN 9780128122006, https://doi.org/10.1016/B978-0-12-801238-3.65333-3. | |||
General references
1. Giugliano RP, Pedersen TR, Park JG, et al. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the FOURIER trial. Lancet 2017;390(10106):1962-1971. doi:10.1016/S0140-6736(17)32290-0
2. Bowe B, Xie Y, Xian H, Balasubramanian S, Zayed MA, Al-Aly Z. High Density Lipoprotein Cholesterol and the Risk of All-Cause Mortality among U.S. Veterans. Clin J Am Soc Nephrol 2016;11(10):1784-1793. doi:10.2215/CJN.00730116
3. Emerging Risk Factors Collaboration, Di Angelantonio E, Sarwar N, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009;302(18):1993-2000. doi:10.1001/jama.2009.1619
4. Isaacs A, Sayed-Tabatabaei FA, Njajou OT, Witteman JC, van Duijn CM. The -514 C->T hepatic lipase promoter region polymorphism and plasma lipids: a meta-analysis. J Clin Endocrinol Metab. 2004;89(8):3858-3863. doi:10.1210/jc.2004-0188
5. Kanai M, Akiyama M, Takahashi A, et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet 2018;50(3):390-400. doi:10.1038/s41588-018-0047-6
6. Madsen CM, Varbo A, Nordestgaard BG. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: two prospective cohort studies. Eur Heart J 2017;38(32):2478-2486. doi:10.1093/eurheartj/ehx163
7. Wilkins JT, Ning H, Stone NJ, et al. Coronary heart disease risks associated with high levels of HDL cholesterol. J Am Heart Assoc 2014;3(2):e000519. Published 2014 Mar 13. doi:10.1161/JAHA.113.000519
8. Yang X, Sethi A, Yanek LR, et al. SCARB1 Gene Variants Are Associated With the Phenotype of Combined High High-Density Lipoprotein Cholesterol and High Lipoprotein (a). Circ Cardiovasc Genet 2016;9(5):408-418. doi:10.1161/CIRCGENETICS.116.001402
9. Mørland JG, Magnus P, Vollset SE, Leon DA, Selmer R, Tverdal A. Associations between serum high-density lipoprotein cholesterol levels and cause-specific mortality in a general population of 345 000 men and women aged 20-79 years. Int J Epidemiol 2023;52(4):1257-1267. doi:10.1093/ije/dyad011
Classification of Dyslipidemia
Dyslipidemias were traditionally classified by patterns of elevation in lipids and lipoproteins (eg, Fredrickson phenotypes), but a more practical system categorizes dyslipidemias as primary or secondary and characterizes them by:
Increases in cholesterol only: Pure or isolated hypercholesterolemia
Increases in triglycerides only: Pure or isolated hypertriglyceridemia
Increases in both cholesterol and triglycerides: Mixed or combined hyperlipidemias
This system does not take into account specific lipoprotein abnormalities (eg, low HDL-C or high LDL-C) that may contribute to disease despite normal cholesterol and triglyceride levels.
Etiology of Dyslipidemia
Dyslipidemias may be:
Primary: Genetic
Secondary: Caused by lifestyle and other factors
Both primary and secondary causes contribute to dyslipidemias in varying degrees. For example, in familial combined hyperlipidemia, expression may occur only in the presence of significant secondary causes.
Primary causes
Primary causes are single or multiple gene mutations that result either in overproduction or defective clearance of triglycerides and LDL or in underproduction or excessive clearance of HDL (see table ).
Genetic (Primary) Dyslipidemias
Disorder | Genetic Defect/Mechanism | Inheritance | Prevalence | Clinical Features | Treatment |
|---|---|---|---|---|---|
Apo C-II deficiency [a] | Apo C-II (causing functional LPL deficiency) | Recessive | < 1/1 million | Pancreatitis (in some adults), metabolic syndrome (often present) TG: > 750 mg/dL (> 8.5 mmol/L) | Diet: Severe fat restriction with fat-soluble vitamin supplementation and medium-chain TG supplementation |
Cerebrotendinous xanthomatosis [b] | Hepatic mitochondrial 27-hydroxylase defect Blockage of bile acid synthesis and conversion of cholesterol to cholestanol, which accumulates in blood, nervous system, and other organs | Recessive | Rare | Cataracts, premature CAD, neuropathy, ataxia | Chenodeoxycholic acid |
Lysosomal acid lipase deficiency | Recessive | Rare | Premature CAD Accumulation of cholesteryl esters and TG in lysosomes in the liver, spleen, and lymph nodes Cirrhosis | Possibly statins Enzyme replacement | |
Familial apo AI deficiency/mutations [d] | Apo AI | Unknown | Rare | Corneal opacities, xanthomas, premature CAD (in some people) HDL: 15–30 mg/dL (0.39–0.78 mmol/L) | Supportive care |
Familial chylomicronemic syndrome (formerly LPL deficiency) [e] | LPL gene APOC2 gene APOA5 gene GP1HBP1 gene LMF1 gene | Recessive | Rare | Hypertriglyceridemia, pancreatitis, eruptive xanthomas, lipemia retinalis TG > 1000 mg/dL (11.2 mmol/L) | Low-fat diet Fibrates Plasma exchange OlezarsenOlezarsen |
Familial combined hyperlipidemia [f] | Unknown, possibly multiple defects and mechanisms | Dominant | 1/50 to 1/100 | Premature CAD, responsible for approximately 15% of MIs in people < 60 years Apo B: Disproportionately elevated TC: 250–500 mg/dL (6.5–13.0 mmol/L) TG: 250–750 mg/dL (2.8–8.5 mmol/L) | Low-fat diet Weight loss Lipid-lowering medications |
Familial defective apo B-100 [g] | Apo B (LDL receptor–binding region defect) Diminished LDL clearance | Dominant | 1/700 | Xanthomas, arcus corneae, premature CAD TC: 250–500 mg/dL (6.5–13 mmol/L) | Low-fat diet Lipid-lowering medications |
Familial dysbetalipoproteinemia [h] | Apo E (usually e2/e2 homozygotes) Diminished chylomicron and VLDL clearance | Recessive (more common) or dominant (less common) | 1/5000 | Xanthomas (especially tuberous and palmar), yellow palmar creases, premature CAD TC: 250–500 mg/dL (6.5–13.0 mmol/L) TG: 250–500 mg/dL (2.8–5.6 mmol/L) | Low-fat diet Lipid-lowering medications |
Familial hypercholesterolemia [i] | LDL receptor defect Diminished LDL clearance | Codominant | Heterozygotes: 1/200 | Tendon xanthomas, arcus corneae, premature CAD (ages 30–50), responsible for approximately 5% of MIs in people < 60 years TC: 250–500 mg/dL (6.5–13 mmol/L) | Low-fat diet Lipid-lowering medications LDL apheresis (for patients who are homozygous and those who are heterozygous with severe disease) |
Homozygotes: 1/250,000–1/1 million (increased among French Canadian, Christian Lebanese, and South African populations) | Planar and tendon xanthomas and tuberous xanthomas, premature CAD (before age 18) TC > 500 mg/dL (> 13 mmol/L) | Low-fat diet Lipid-lowering medications LDL apheresis (for patients who are homozygous and those who are heterozygous with severe disease) Liver transplantation (for homozygous patients) | |||
Familial hypertriglyceridemia [h] | Unknown, possibly multiple defects and mechanisms | Dominant | 1/500 | Usually no symptoms or findings; occasionally hyperuricemia, sometimes early atherosclerosis TG: 200–500 mg/dL (2.3–5.6 mmol/L), possibly higher depending on diet and alcohol use | Low-fat diet Weight loss Lipid-lowering medications |
Familial LCAT deficiency [j] | LCAT gene | Recessive | Extremely rare | Corneal opacities, anemia, chronic kidney disease HDL: < 10 mg/dL (< 0.26 mmol/L) | Fat restriction Renal transplantation |
Fisheye disease (partial LCAT deficiency) | LCAT gene | Recessive | Extremely rare | Corneal opacities HDL: < 10 mg/dL (< 0.26 mmol/L) | Supportive care |
Hepatic lipase deficiency | Hepatic lipase | Recessive | Extremely rare | Premature CAD TC: 250–1500 mg/dL (6.5–39 mmol/L) TG: 395–8200 mg/dL (4.5–93 mmol/L) HDL: Variable | Low-fat diet, lipid-lowering medications |
PCSK9 gain of function mutations | Increased degradation of LDL receptors | Dominant | Unknown | Similar to familial hypercholesterolemia | Low-fat diet Lipid-lowering medications |
Polygenic hypercholesterolemia | Unknown, possibly multiple defects and mechanisms | Variable | Common | Premature CAD TC: 250–350 mg/dL (6.5–9.0 mmol/L) | Low-fat diet Lipid-lowering medications |
Primary hypoalphalipoproteinemia (familial or nonfamilial) | Unknown, possibly apo A-I, C-III, or A-IV | Dominant | Rare | Premature CAD HDL: 15–35 mg/dL (0.39–0.91 mmol/L) | Exercise LDL-lowering medications |
Sitosterolemia | ABCG5 and ABCG8 genes | Recessive | Rare | Tendon xanthomas, premature CAD | Fat restriction Bile acid sequestrants EzetimibeEzetimibe |
Tangier disease | ABCA1 gene | Recessive | Rare | Premature CAD (in some people), peripheral neuropathy, hemolytic anemia, corneal opacities, hepatosplenomegaly, orange tonsils HDL: < 5 mg/dL (< 0.13 mmol/L) | Low-fat diet |
[a] Prevalence data from Hoffmann, M.M., März, W. (2009). Apo C-II Deficiency. In: Lang, F. (eds) Encyclopedia of Molecular Mechanisms of Disease. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-29676-8_137 | |||||
[b] Prevalence data from Nie S, Chen G, Cao X, Zhang Y. Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J Rare Dis 2014;9:179. Published 2014 Nov 26. doi:10.1186/s13023-014-0179-4 | |||||
[c] Prevalence data from Pericleous M, Kelly C, Wang T, Livingstone C, Ala A. Wolman's disease and cholesteryl ester storage disorder: the phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol Hepatol 2017;2(9):670-679. doi:10.1016/S2468-1253(17)30052-3 | |||||
[d] Data from Geller AS, Polisecki EY, Diffenderfer MR, et al. Genetic and secondary causes of severe HDL deficiency and cardiovascular disease. J Lipid Res 2018;59(12):2421-2435. doi:10.1194/jlr.M088203 | |||||
[e] Prevalence data from Javed F, Hegele RA, Garg A, et al. Familial chylomicronemia syndrome: An expert clinical review from the National Lipid Association. J Clin Lipidol published March 21, 2025. doi: 10.1016/j.jacl.2025.03.013 | |||||
[f] Prevalence data from Taghizadeh E, Farahani N, Mardani R, Taheri F, Taghizadeh H, Gheibihayat SM. Genetics of Familial Combined Hyperlipidemia (FCHL) Disorder: An Update. Biochem Genet 2022;60(2):453-481. doi:10.1007/s10528-021-10130-2 | |||||
[g] Prevalence data from Tybjaerg-Hansen A, Humphries SEAtherosclerosis. Familial defective apolipoprotein B-100: a single mutation that causes hypercholesterolemia and premature coronary artery disease. 1992;96(2-3):91-107. doi:10.1016/0021-9150(92)90056-m | |||||
[h] Data from Shah AS, Wilson DP. Genetic Disorders Causing Hypertriglyceridemia in Children and Adolescents. [Updated 2023 Feb 22]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK395571/ | |||||
[i] Data from Jackson CL, Ahmad Z, Das SR, Khera A. The evaluation and management of patients with LDL-C ≥ 190 mg/dL in a large health care system. Am J Prev Cardiol 2020;1:100002. Published 2020 May 1. doi:10.1016/j.ajpc.2020.100002 | |||||
[j] Data from Mehta R, Elias-Lopez D, Martagon AJ, et al. LCAT deficiency: a systematic review with the clinical and genetic description of Mexican kindred. Lipids Health Dis 2021;20(1):70. Published 2021 Jul 13. doi:10.1186/s12944-021-01498-6 | |||||
ABCA1 =adenosine triphosphate (ATP)-binding cassette transporter A1; ABCG5 and 8 = ATP-binding cassette subfamily G members 5 and 8; apo = apoprotein; CAD = coronary artery disease; HDL = high-density lipoprotein; LCAT =lecithin-cholesterol acyltransferase; LDL = low-density lipoprotein; LPL = lipoprotein lipase; MI = myocardial infarction; PCSK9 = proprotein convertase subtilisin-like/kexin type 9; TC = total cholesterol; TG = triglyceride; VLDL = very-low-density lipoprotein. | |||||
Secondary causes
Secondary causes contribute to many cases of dyslipidemia in adults.
The most important secondary cause of dyslipidemia in high-resource countries is:
A sedentary lifestyle with excessive dietary intake of total calories, saturated fat, cholesterol, and trans fats
Trans fats are polyunsaturated or monounsaturated fatty acids to which hydrogen atoms have been added; they are used in some processed foods and are as atherogenic as saturated fat.
Other common secondary causes of dyslipidemia include:
Primary biliary cirrhosis and other cholestatic liver diseases
Medications
Medications, such as thiazides, beta-blockers, retinoids, highly active antiretroviral agents, cyclosporine, tacrolimus, progestins, and glucocorticoids can cause secondary dyslipidemia. Oral Medications, such as thiazides, beta-blockers, retinoids, highly active antiretroviral agents, cyclosporine, tacrolimus, progestins, and glucocorticoids can cause secondary dyslipidemia. Oralestrogens cause a mixed effect (decrease LDL-C and increase HDL-C, but also increase triglycerides).
Secondary causes of low levels of HDL-C include cigarette smoking, anabolic steroids, HIV infection, and nephrotic syndrome. Data on the effects of smokeless tobacco and marijuana on lipid metabolism are sparse (1, 2).
Diabetes is an especially significant secondary cause because patients tend to have an atherogenic combination of high triglycerides; high small, dense LDL fractions; and low HDL (diabetic dyslipidemia, hypertriglyceridemic hyperapo B). Patients with type 2 diabetes are especially at risk. The combination may be a consequence of obesity, poor control of diabetes, or both, which may increase circulating free fatty acids (FFAs), leading to increased hepatic very-low-density lipoprotein (VLDL) production. triglyceride-rich VLDL then transfers triglyceride and cholesterol to LDL and HDL, promoting formation of triglyceride-rich, small, dense LDL and clearance of triglyceride-rich HDL. Diabetic dyslipidemia is often exacerbated by the increased caloric intake and physical inactivity that characterize the lifestyle of some patients with type 2 diabetes. Women with diabetes may be at special risk of cardiac disease as a result of this form of dyslipidemia.
Etiology references
1. Ravi D, Ghasemiesfe M, Korenstein D, Cascino T, Keyhani S. Associations Between Marijuana Use and Cardiovascular Risk Factors and Outcomes: A Systematic Review. Ann Intern Med 2018;168(3):187-194. doi:10.7326/M17-1548
2. Tucker LA. Use of smokeless tobacco, cigarette smoking, and hypercholesterolemia. Am J Public Health 1989;79(8):1048-1050. doi:10.2105/ajph.79.8.1048
Symptoms and Signs of Dyslipidemia
Dyslipidemia itself usually causes no symptoms, although very high triglyceride levels can cause paresthesias, dyspnea, and confusion. Lipid disorders can lead to symptomatic end-organ disease, including:
Vascular disease (eg, coronary artery disease [CAD], stroke, peripheral arterial disease)
Acute pancreatitis can be caused be high triglyceride levels (> 500 mg/dL [> 5.65 mmol/L])
Hepatosplenomegaly can be caused by very high triglyceride levels (> 500 mg/dL [> 5.65 mmol/L])
Findings in patients with severe dyslipidemia (LDL > 190 mg/dL [> 4.9 mmol/L]) may include localized lipid deposits (xanthomas) or other findings caused by high serum concentrations or accumulation of lipids.
High LDL-C levels can cause tendinous xanthomas at the Achilles, elbow, and knee tendons and over metacarpophalangeal joints. Other clinical findings that occur in patients with high LDL-C (eg, in familial hypercholesterolemia or dysbetalipoproteinemia) include planar or tuberous xanthomas. Planar xanthomas are flat or slightly raised yellowish patches. Tuberous xanthomas are painless, firm nodules typically located over extensor surfaces of joints.
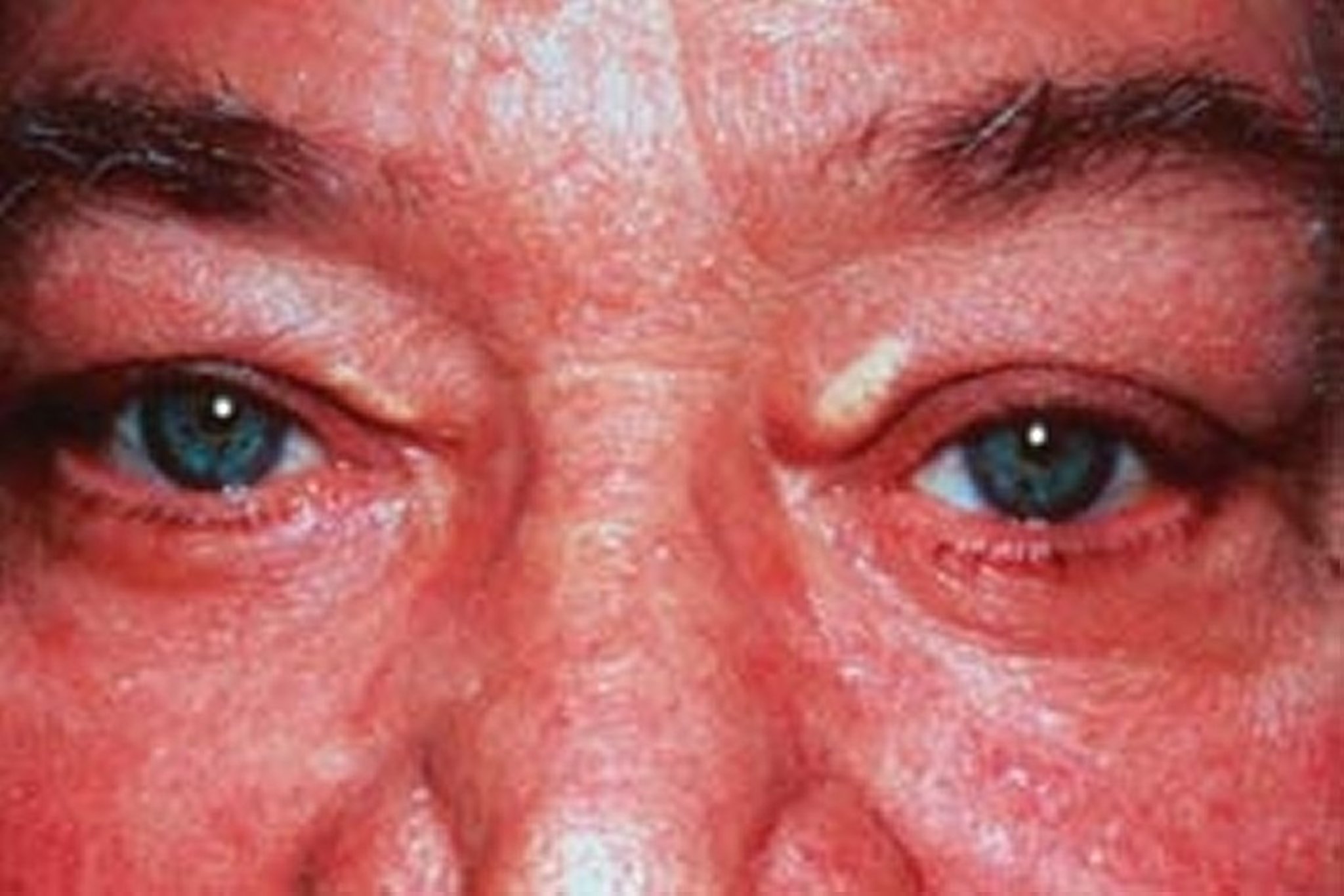
Patients with high levels of LDL-C can develop arcus corneae (lipid deposits in the cornea around the iris) and xanthelasma (lipid-rich, yellow plaques on the medial eyelids). Xanthelasma can also occur in patients with primary biliary cirrhosis and normal lipid levels.
Severe elevations of triglycerides can cause eruptive xanthomas over the trunk, back, elbows, buttocks, knees, hands, and feet. Severe hypertriglyceridemia (> 2000 mg/dL [> 22.6 mmol/L]) can also give retinal arteries and veins a creamy white appearance (lipemia retinalis).

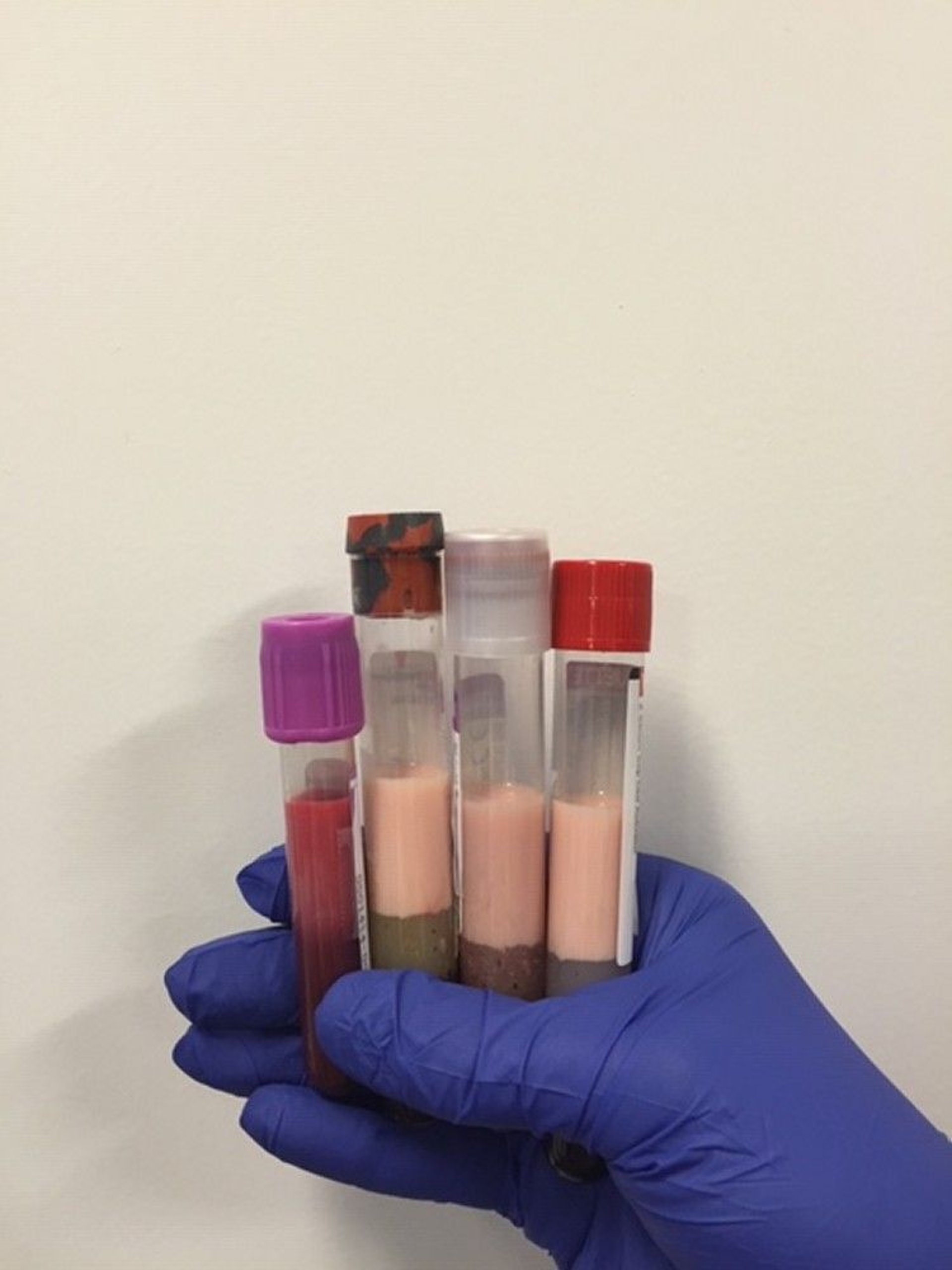
Image courtesy of Michael H. Davidson, MD.

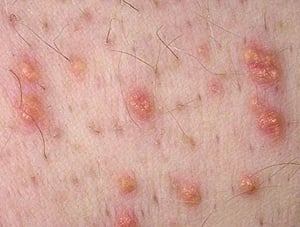
Eruptive xanthomas are skin manifestations of elevated triglyceride levels.
Eruptive xanthomas are skin manifestations of elevated triglyceride levels.
Photo provided by Thomas Habif, MD.

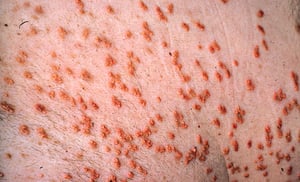
Patients with severe elevations of triglycerides can have eruptive xanthomas over the trunk, back, elbows, buttocks, knees, hands, and feet.
Patients with severe elevations of triglycerides can have eruptive xanthomas over the trunk, back, elbows, buttocks, kn
© Springer Science+Business Media

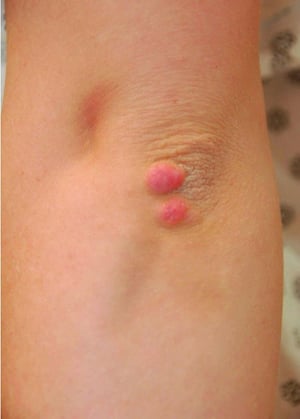
Tuberous xanthomas are painless, firm nodules typically located over extensor surfaces of joints.
Tuberous xanthomas are painless, firm nodules typically located over extensor surfaces of joints.
© Springer Science+Business Media

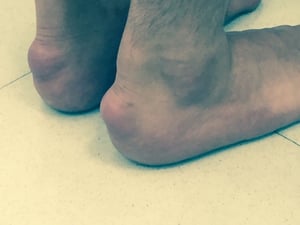
Achilles tendon xanthomas are diagnostic of familial hypercholesterolemia.
Achilles tendon xanthomas are diagnostic of familial hypercholesterolemia.
Image courtesy of Michael H. Davidson, MD.
Tendon xanthomas (arrow) are diagnostic of familial hypercholesterolemia.
Tendon xanthomas (arrow) are diagnostic of familial hypercholesterolemia.
Image courtesy of Michael H. Davidson, MD.
Eruptive xanthomas are skin manifestations of elevated triglyceride levels.
Eruptive xanthomas are skin manifestations of elevated triglyceride levels.
Photo provided by Thomas Habif, MD.
Patients with severe elevations of triglycerides can have eruptive xanthomas over the trunk, back, elbows, buttocks, knees, hands, and feet.
Patients with severe elevations of triglycerides can have eruptive xanthomas over the trunk, back, elbows, buttocks, kn
© Springer Science+Business Media
Tuberous xanthomas are painless, firm nodules typically located over extensor surfaces of joints.
Tuberous xanthomas are painless, firm nodules typically located over extensor surfaces of joints.
© Springer Science+Business Media
Achilles tendon xanthomas are diagnostic of familial hypercholesterolemia.
Achilles tendon xanthomas are diagnostic of familial hypercholesterolemia.
Image courtesy of Michael H. Davidson, MD.
Tendon xanthomas (arrow) are diagnostic of familial hypercholesterolemia.
Tendon xanthomas (arrow) are diagnostic of familial hypercholesterolemia.
Image courtesy of Michael H. Davidson, MD.
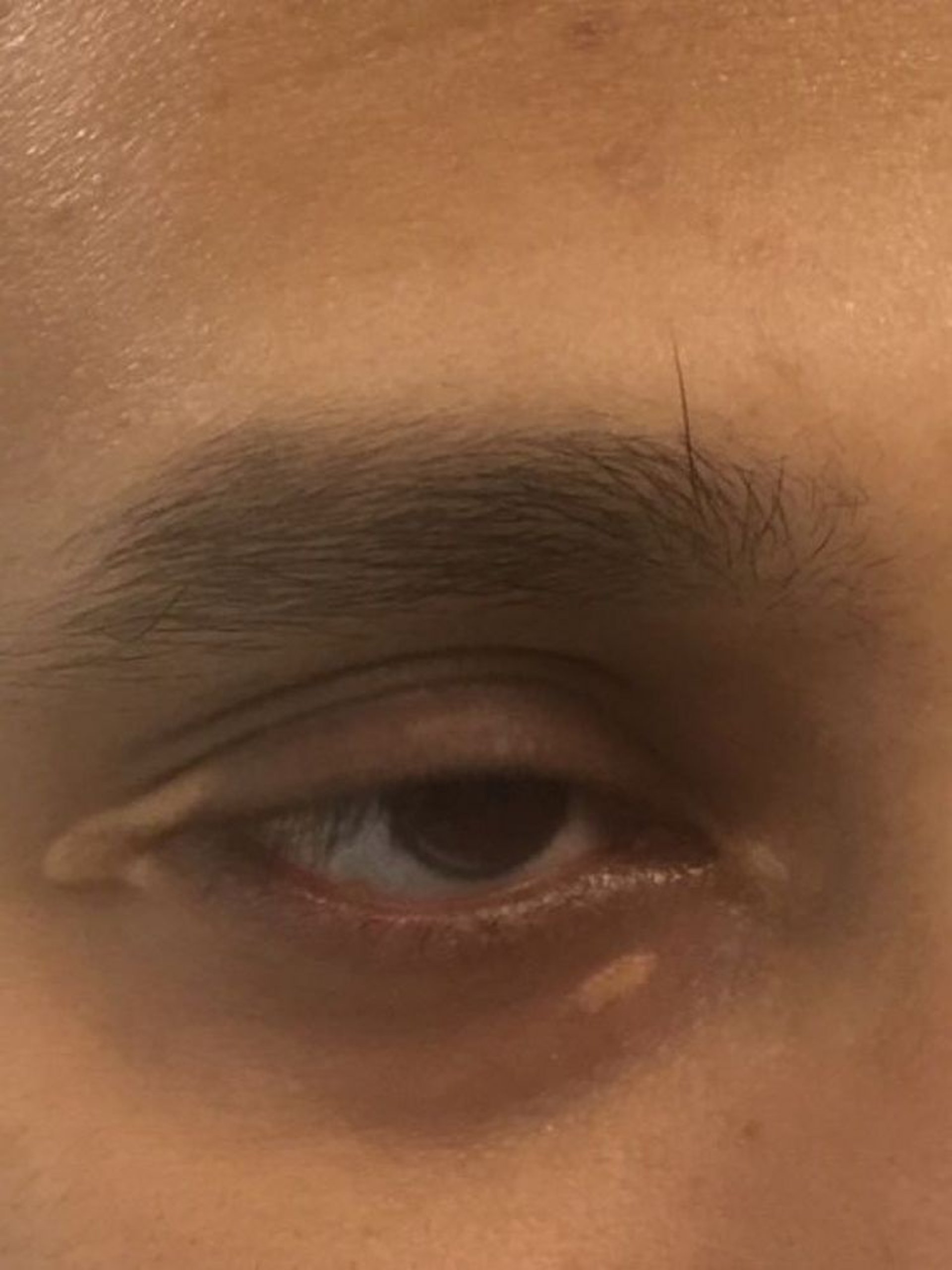
Image courtesy of Michael H. Davidson, MD.

© Springer Science+Business Media
Diagnosis of Dyslipidemia
Serum lipid profile (measured total cholesterol, triglyceride, and HDL-C and calculated LDL-C and VLDL-C)
Dyslipidemia is diagnosed by measuring serum lipids. Routine measurements (lipid profile) include total cholesterol (TC), triglycerides, HDL-C, and LDL-C; these results are used to calculate LDL-C and VLDL-C.
Dyslipidemia is often diagnosed with routine screening tests. It may also be suspected in patients with complications of dyslipidemia (eg, atherosclerotic disease). Physical findings are less common, and suggest primary dyslipidemia.
Primary lipid disorders are suspected when patients have:
Physical signs of dyslipidemia, such as tendon xanthomas, which are pathognomonic for familial hypercholesterolemia
Onset of premature atherosclerotic disease (men < 55 years, women < 60 years)
Family history of premature atherosclerotic disease or severe hyperlipidemia
Serum LDL-C > 190 mg/dL (> 4.9 mmol/L)
Lipid profile measurement
Total cholesterol (TC), triglycerides (TGs), and HDL-C are measured directly. TC and triglyceride values reflect cholesterol and triglycerides in all circulating lipoproteins, including chylomicrons, VLDL, intermediate-density lipoprotein (IDL), LDL, and HDL. TC values can vary by 10% and triglycerides by up to 25% day-to-day even in the absence of a disorder.
Clinical practice has previously been to measure lipids in a fasting state; however, guideline changes support screening most patients in a non-fasting state to support patient adherence to testing. Most experts recommend fasting lipids only in specific situations, including when triglycerides are > 400 mg/dL (> 4.5 mmol/L), which may be associated with secondary causes of hypertriglyceridemia (1, 2).
Pearls & Pitfalls
|
Testing should be postponed if a patient has an acute illness, because triglyceride and lipoprotein(a) levels increase and cholesterol levels decrease in inflammatory states. Also, lipid profiles can vary for about 30 days after an acute myocardial infarction (MI); however, results obtained within 24 hours after MI are usually reliable enough to guide initial lipid-lowering therapy.
LDL-C values are most often calculated as the amount of cholesterol not contained in HDL and VLDL. VLDL is estimated by triglyceride ÷ 5 because the cholesterol concentration in VLDL particles is usually one-fifth of the total lipid in the particle. Thus, the Friedewald formula estimates LDL-C as follows:

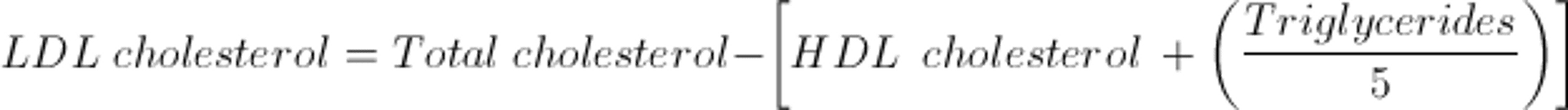
The calculated LDL cholesterol value incorporates measures of all non-HDL and nonchylomicron cholesterol, including that in IDL and lipoprotein (a) (Lp(a)).
For patients who have elevated triglyceride levels, errors in estimating VLDL-C are magnified while using the constant factor of 5. The Martin-Hopkins equation may be used to obtain a more reliable estimate of LDL-C. The constant factor of 5 is replaced with a novel factor based on a patient's non-HDL-C and triglyceride values (3). The novel factor is an adjustable factor that is derived from a table. Both the Friedewald and Martin-Hopkins equations were developed and validated for fasting patients with serum triglyceride levels < 400 mg/dL (< 4.5 mmol/L).
The Martin-Hopkins equation is as follows:

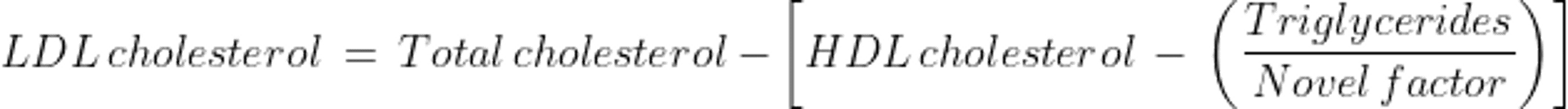
LDL-C can also be measured directly using plasma ultracentrifugation, which separates chylomicrons and VLDL fractions from HDL-C and LDL-C, and by an immunoassay method. Direct measurement may be useful in some patients with elevated triglycerides, but these direct measurements are not usually necessary.

Other tests
In some patients, additional lipid tests should be done.
Lp(a) levels and C-reactive protein levels should be measured in patients with premature atherosclerotic cardiovascular disease, cardiovascular disease (even if they have lower risk lipid levels; see table ), or high LDL-C levels refractory to pharmacotherapy. Some guidelines recommend that Lp(a) levels be checked at least once in all adults and in children who have had ischemic strokes or who have a family history of premature coronary disease (4). Lp(a) levels may also be directly measured in patients with borderline-high LDL-C levels to determine whether pharmacotherapy is warranted.
Measurements of LDL particle number or apoprotein B-100 (apo B) is useful in patients with elevated triglycerides and the metabolic syndrome. Apo B provides similar information to LDL particle number because there is one apo B molecule for each LDL particle. Apo B measurement includes all atherogenic particles, including remnants and Lp(a).
Apo B value reflects all non-HDL-C (in VLDL, IDL, and LDL) and is more predictive of CAD risk than LDL-C. Non-HDL-C (TC − HDL-C) is also more predictive of CAD risk than LDL-C, especially in patients with hypertriglyceridemia.
Secondary causes
Tests for secondary causes of dyslipidemia should be done in most patients with newly diagnosed dyslipidemia and repeated when a component of the lipid profile has inexplicably changed for the worse. Such tests include measurements of
Creatinine
Fasting glucose and/or glycosylated hemoglobin (HbA1C)
Liver enzymes
Thyroid-stimulating hormone (TSH)
Urinary protein
Diagnosis references
1. Nordestgaard BG, Langsted A, Mora S, et al. Fasting is not routinely required for determination of a lipid profile: clinical and laboratory implications including flagging at desirable concentration cut-points-a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur Heart J 2016;37(25):1944-1958. doi:10.1093/eurheartj/ehw152
2. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines [published correction appears in Circulation 2019 Jun 18;139(25):e1182-e1186. doi: 10.1161/CIR.0000000000000698.] [published correction appears in Circulation 2023 Aug 15;148(7):e5. doi: 10.1161/CIR.0000000000001172.]. Circulation 2019;139(25):e1082-e1143. doi:10.1161/CIR.0000000000000625
3. Martin SS, Blaha MJ, Elshazly MB, et al. Comparison of a novel method vs the Friedewald equation for estimating low-density lipoprotein cholesterol levels from the standard lipid profile. JAMA 310(19):2061-2068, 2013. doi:10.1001/jama.2013.280532
4. Kronenberg F, Mora S, Stroes ESG, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J 2022;43(39):3925-3946. doi:10.1093/eurheartj/ehac361
Screening for Dyslipidemia
Screening is done using a fasting lipid profile (TC, triglycerides, HDL-C, and calculated LDL-C). Medical society guidelines vary regarding when to begin screening. Based on the risk factors, screening can begin as early as age 2 in children with family history of heart disease or familial hypercholesterolemia.
Lipid measurement should be accompanied by assessment for other cardiovascular risk factors, including:
Cigarette use
Family history of CAD in a male first-degree relative before age 55 years or a female first-degree relative before age 65 years
Screening in children
Most physicians recommend screening per the 2012 National Heart Lung and Blood Institute Guidelines (1) as follows:
Children with risk factors (eg, diabetes, hypertension, family history of severe hyperlipidemia or premature CAD): Fasting lipid profile once at age 2 to 8
Children with no risk factors: Non-fasting or fasting lipid profile once before puberty (usually age 9 to 11) and once more at age 17 to 21 (2)
Screening in adults
Adults should be screened at age 20 (3, 4) and every 5 years thereafter.
An age to discontinue screening has not been established, but evidence supports screening of patients into their 80s, especially if they have atherosclerotic cardiovascular disease (5).
Patients with an extensive family history of heart disease—heart attack, stroke, or coronary artery disease before age 55 (in men) or age 65 (in women) without known risk factors, such as high LDL, smoking, diabetes, or obesity, or known family history of high Lp(a)—should also be screened by measuring Lp(a) levels.
Screening references
1. Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents. 2011.
2. Daniels SR, Greer FR; Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008;122(1):198-208. doi:10.1542/peds.2008-1349
3. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al: 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk. J Am Coll Cardiol 63:2935–2959, 2014. doi: 10.1016/j.jacc.2013.11.005
4. Grundy SM, Stone NJ, Bailey AL, et al: 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 139: e1082–e1143, 2019. doi: 10.1161/CIR.0000000000000625
5. Kleipool EE, Dorresteijn JA, Smulders YM, et al: Treatment of hypercholesterolaemia in older adults calls for a patient-centred approach. Heart 106(4):261-266, 2020. doi:10.1136/heartjnl-2019-315600
Treatment of Dyslipidemia
Lifestyle changes (eg, exercise, dietary modification)
For high LDL-C, statins, bile acid sequestrants, ezetimibe, bempedoic acid, and PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitorsFor high LDL-C, statins, bile acid sequestrants, ezetimibe, bempedoic acid, and PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitors
For high triglyceride, fibrates, omega-3 fatty acids, and sometimes other measures
General principles
The main goal for dyslipidemia treatment is prevention of atherosclerotic cardiovascular disease (ASCVD), including acute coronary syndromes, stroke, transient ischemic attack, or peripheral artery disease presumed caused by atherosclerosis. Treatment is indicated for all patients with ASCVD (secondary prevention) and for some without (primary prevention).
The American Academy of Pediatrics (AAP) recommends treatment for some children who have elevated LDL-C levels (1). Children with heterozygous familial hypercholesterolemia should be treated beginning at age 8 to 10 years. Children with homozygous familial hypercholesterolemia require diet, medications, and often LDL apheresis to prevent premature death; treatment is begun when the diagnosis is made.
Treatment options depend on the specific lipid abnormality, although different lipid abnormalities often coexist. In some patients, a single abnormality may require several therapies; in others, a single treatment may be adequate for several abnormalities. Treatment should always include treatment of hypertension and diabetes and smoking cessation. Treatment should also include daily low-dose aspirin in patients age 40 to 79 years with a low risk of bleeding and with a 10-year risk of myocardial infarction or death due to CAD of ≥ 20%. In general, treatment options for men and women are the same.
Elevated LDL-C treatment
For all individuals, the prevention of ASCVD requires an emphasis on a heart-healthy lifestyle, particularly diet and exercise. Other options to lower LDL-C in all age groups include medications, dietary supplements, and procedural interventions. Many of these options are also effective for treating other lipid abnormalities.
Dietary changes help to maintain ideal body weight and provide other benefits. These changes include:
Decreasing intake of saturated fats
Increasing the amount of dietary soluble fiber (eg, oatmeal, oat bran, psyllium)Increasing the amount of dietary soluble fiber (eg, oatmeal, oat bran, psyllium)
Response to dietary changes can be variable, and lipid profile should be reassessed after a nutritional intervention to determine its effectiveness. Referral to a dietitian is often useful.
Exercise lowers LDL-C and raises HDL-C in some people (2) and also helps maintain ideal body weight. The American Heart Association (AHA) recommends 150 minutes of moderate-intensity exercise or 75 minutes of high-intensity exercise weekly, as well as 2 moderate-intensity strength training sessions weekly (3).
Dietary changes and exercise should be used whenever feasible, but AHA/American College of Cardiology (ACC) guidelines recommend also using pharmacotherapy for certain groups of patients after discussion of the risks and benefits of statin therapy (4).
For pharmacotherapy in adults, the AHA/ACC Guidelines recommend treatment with a statin for 4 groups of patients, comprised of those with any of the following:
Clinical ASCVD
LDL-C ≥ 190 mg/dL (≥ 4.9 mmol/L)
Age 40 to 75 years, with diabetes and LDL-C 70 to 189 mg/dL (1.8 to 4.9 mmol/L)
Age 40 to 75 years, with LDL-C 70 to 189 mg/dL (1.8 to 4.9 mmol/L), and an estimated 10-year risk of ASCVD ≥ 7.5%
The standard tool to quantify cardiovascular risk is the pooled cohort risk assessment equation, which was validated by a 2013 joint ACC/AHA work group. This cardiovascular risk calculator is based on sex, age, race, total and HDL-C, systolic and diastolic blood pressure, diabetes, smoking status, and the use of antihypertensives and statins (5).
More recently, the AHA has introduced the PREVENT risk calculator for use in primary prevention in patients without coronary heart disease, stroke or heart failure (6). It expands the age range which can be stratified and incorporates hemoglobin A1C, urine albumin-to-creatinine ratio, and the social deprivation index (which uses zip code to estimate deprivation based on seven demographic characteristics). Increased lifetime risk (identified using the PREVENT risk calculator) is relevant because 10-year risk may be low in younger patients, in whom longer-term risk should be taken into account.
When considering whether to give a statin, clinicians may also take into account other factors, including the following:
LDL-C ≥ 160 mg/dL (4.1 mmol/L)
Family history of premature ASCVD (ie, age of onset < 55 years in a male first-degree relative, or < 65 years in a female first-degree relative)
High-sensitivity C-reactive protein ≥ 2 mg/L (≥ 19 nmol/L)
Coronary artery calcium score ≥ 300 Agatston units (or ≥ 75th percentile for the patient's demographic)
Ankle-brachial index < 0.9
Increased lifetime risk
ACC/AHA guidelines expand this list of risk enhancing factors to include the following (7):
Family history of premature ASCVD (ie, age of onset in males age < 55 years or females age < 65 years)
South Asian ancestry
Female-specific risk factors (history of preeclampsia or premature menopause)
Persistently elevated LDL-C (> 160 mg/dL [4.1 mmol/L])
Persistently elevated triglycerides (> 175 mg/dL [1.98 mmol/L])
Chronic kidney disease
Metabolic syndrome
Inflammatory disease (especially rheumatoid arthritis, HIV, psoriasis)
Elevated Lp(a) (> 50 mg/dL or 125 nmol/L)
Elevated apolipoprotein B (Apo B) (> 130 mg/dL [1.30 g/L])
Ankle-brachial index (ABI) < 0.9
Statins are the treatment of choice for LDL-C reduction because evidence has demonstrated that they reduce cardiovascular morbidity and mortality (8). Other classes of lipid-lowering medications are not the first choice for treating elevated LDL-C because they have not demonstrated equivalent efficacy for decreasing ASCVD.
Statins inhibit hydroxymethylglutaryl CoA reductase, a key enzyme in cholesterol synthesis, leading to up-regulation of LDL receptors and increased LDL clearance. They reduce LDL-C by up to 60% and produce small increases in HDL-C and modest decreases in triglycerides. Statins also appear to decrease intra-arterial inflammation, systemic inflammation, or both by stimulating production of endothelial nitric oxide and may have other beneficial effects.Statins inhibit hydroxymethylglutaryl CoA reductase, a key enzyme in cholesterol synthesis, leading to up-regulation of LDL receptors and increased LDL clearance. They reduce LDL-C by up to 60% and produce small increases in HDL-C and modest decreases in triglycerides. Statins also appear to decrease intra-arterial inflammation, systemic inflammation, or both by stimulating production of endothelial nitric oxide and may have other beneficial effects.
Statin therapy is classified as high-, moderate-, or low-intensity and is given based on treatment group and age (see table ). The choice of statin depends on the patient's co-morbidities, other medications, risk factors for adverse events, statin intolerance, cost, and patient preference.
Adverse effects with statins are uncommon but include liver enzyme elevations and myositis or rhabdomyolysis. Liver enzyme elevations are uncommon, and serious liver toxicity is extremely rare. Symptoms or severe adverse effects involving muscle occur in up to 10% of patients taking statins and may be dose-dependent. Muscle symptoms can occur without muscle enzyme (eg, creatine kinase) elevation. Adverse effects are more common among older patients, patients with several disorders, and patients taking several medications. In some patients, changing from one statin to another or lowering the dose (after temporarily discontinuing the medication) relieves the problem. Muscle toxicity seems to be most common when some of the statins are used with medications that inhibit cytochrome P3A4 (eg, macrolide antibiotics, azole antifungals, cyclosporine) and with fibrates, especially gemfibrozil. Statins are contraindicated during pregnancy and lactation.. Liver enzyme elevations are uncommon, and serious liver toxicity is extremely rare. Symptoms or severe adverse effects involving muscle occur in up to 10% of patients taking statins and may be dose-dependent. Muscle symptoms can occur without muscle enzyme (eg, creatine kinase) elevation. Adverse effects are more common among older patients, patients with several disorders, and patients taking several medications. In some patients, changing from one statin to another or lowering the dose (after temporarily discontinuing the medication) relieves the problem. Muscle toxicity seems to be most common when some of the statins are used with medications that inhibit cytochrome P3A4 (eg, macrolide antibiotics, azole antifungals, cyclosporine) and with fibrates, especially gemfibrozil. Statins are contraindicated during pregnancy and lactation.
In patients with ASCVD, risk reduction increases with decreasing LDL-C levels. Thus, initial treatment is a statin at maximally tolerated dose with the goal of lowering LDL-C by > 50% (high-intensity therapy). For very high-risk patients with ASCVD (eg, those with a recent myocardial infarction or unstable angina or with high-risk comorbidities such as diabetes), a LDL-C level > 70 mg/dL (> 1.2 mmol/L) despite maximal statin therapy should prompt the addition of ezetimibe or a PCSK9 inhibitor (eg, evolocumab, alirocumab). These therapies have been proven to reduce major adverse cardiovascular events in conjunction with statin therapy in large clinical outcome trials (), a LDL-C level > 70 mg/dL (> 1.2 mmol/L) despite maximal statin therapy should prompt the addition of ezetimibe or a PCSK9 inhibitor (eg, evolocumab, alirocumab). These therapies have been proven to reduce major adverse cardiovascular events in conjunction with statin therapy in large clinical outcome trials (9, 10).
Non-statin medications also have a place in lowering LDL-C (see table ). The 2022 ACC Expert Consensus Decision Pathway on the Role of Non-statin Therapies provides guidance to clinicians on the role of non-statin therapy in both primary and secondary prevention of ASCVD (11). Statins are generally first-line medications, but in patients with statin intolerance, or those who are not able to achieve target LDL-C levels despite statin use, non-statin medications are used.
Statins For ASCVD Prevention
Classification | Effects* | Recommended For | Examples† |
|---|---|---|---|
High-intensity | Lowers LDL-C ≥ 50% | Clinical ASCVD, age ≤ 75 years LDL-C ≥ 190 mg/dL (4.9 mmol/L) Age 40 to 75 years, 10-year ASCVD risk ≥ 7.5% Diabetes and 10-year ASCVD risk ≥ 7.5% | Atorvastatin 40-80 mgAtorvastatin 40-80 mg Rosuvastatin 20-40 mgRosuvastatin 20-40 mg |
Moderate-intensity | Lowers LDL-C 30 to < 50% | Clinical ASCVD, age > 75 years Age 40 to 75 years, 10-year ASCVD risk 5 to < 7.5% Age 40 to 75 years, 10-year ASCVD risk ≥ 7.5% (if unable to tolerate high-intensity statin) Diabetes and 10-year ASCVD risk < 7.5% | Atorvastatin 10-20 mgAtorvastatin 10-20 mg Fluvastatin XL 80 mgFluvastatin XL 80 mg Lovastatin 40 mgLovastatin 40 mg Pitavastatin 2-4 mgPitavastatin 2-4 mg Pravastatin 40-80 mgPravastatin 40-80 mg Rosuvastatin 5-10 mgRosuvastatin 5-10 mg Simvastatin 20-40 mgSimvastatin 20-40 mg |
Low-intensity | Lowers LDL-C < 30% | Patients who cannot tolerate high or moderate intensity treatment | Fluvastatin 20-40 mgFluvastatin 20-40 mg Lovastatin 20 mgLovastatin 20 mg Pitavastatin 1 mgPitavastatin 1 mg Pravastatin 10-20 mgPravastatin 10-20 mg |
* Individual response may vary. | |||
† All doses are oral and given once daily. | |||
ASCVD = atherosclerotic cardiovascular disease; LDL-C = low density lipoprotein cholesterol. | |||
Data from Grundy SM, Stone NJ, Bailey AL, et al: 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 139: e1082–e1143, 2019. doi: 10.1161/CIR.0000000000000625 | |||
Non-Statin Lipid–Lowering Drugs*
Medication | Comments |
|---|---|
Adenosine triphosphate citrate lyase inhibitor | Lowers LDL-C |
Bempedoic acidBempedoic acid | Risk of hyperuricemia, tendon rupture Especially useful in patients with statin-associated muscle adverse effects as the enzyme required to activate this drug is absent in the muscle |
Bile acid sequestrants | Lower LDL-C (primary), slightly increase HDL-C (secondary), may increase TGs |
CholestyramineCholestyramine | — |
ColesevelamColesevelam | — |
ColestipolColestipol | — |
Cholesterol absorption inhibitor | Lowers LDL-C (primary), minimally increases HDL-C |
EzetimibeEzetimibe | — |
Medications for homozygous familial hypercholesteremia | Lower LDL-C |
EvinacumabEvinacumab | Given as an intravenous infusion over 60 minutes Adverse reactions in clinical trials include nasopharyngitis, influenza-like illness, and infusion reaction |
LomitapideLomitapide | Risk of hepatotoxicity Increase dose gradually (about every 2 weeks) Measure transaminase levels before increasing dosage |
Fibrates | Lower TGs and VLDL, increase HDL-C, may increase LDL-C (in patients with high TGs) |
Bezafibrate | Decreased dose required in renal insufficiency Not available in the United States |
Ciprofibrate | Not available in the United States |
FenofibrateFenofibrate | Decreased dose required in renal insufficiency May be safest fibrate for use with statins |
GemfibrozilGemfibrozil | Decreased dose required in renal insufficiency |
Nicotinic acid (niacin)Nicotinic acid (niacin) | |
Increases HDL-C; lowers TGs (low doses), LDL-C (higher doses), and Lp(a) (secondary) Frequent adverse effects: Flushing, impaired glucose tolerance, increased uric acid Aspirin and administration with food minimize flushing | |
PCSK9 monoclonal antibodies | Lower LDL-C and TG, may lower Lp(a) |
AlirocumabAlirocumab | For patients with familial hypercholesterolemia and for those with other high-risk concerns |
EvolocumabEvolocumab | For patients with familial hypercholesterolemia and for those with other high-risk concern |
Prescription omega-3 fatty acids | |
Eicosapentaenoic acid ethyl ester (EPA) + Docosahexaenoic acid (DHA) | Lowers TGs Increases LDL-C |
Eicosapentaenoic acid ethyl ester (EPA) only | Lowers TGs only |
SiRNA targeting PCSK9 | Lowers LDL-C and TG, may lower LP(a) |
InclisiranInclisiran | For patients with familial hypercholesterolemia and for other high-risk patients |
* Many of these medications are also available in combination with a statin. | |
HDL = high-density lipoprotein; HDL-C = HDL cholesterol; LDL = low-density lipoprotein; LDL-C = LDL cholesterol; Lp(a) = lipoprotein (a); PCSK9 = proprotein convertase subtilisin-like/kexin type 9; SiRNA = small interfering ribonucleic acid; TG = triglyceride; VLDL = very low-density lipoprotein. | |
The adenosine triphosphate citrate lyase inhibitor, bempedoic acid impairs cholesterol synthesis in the liver and increases LDL receptors. It lowers LDL-C by 15 to 17% (bempedoic acid impairs cholesterol synthesis in the liver and increases LDL receptors. It lowers LDL-C by 15 to 17% (12, 13). Bempedoic acid is especially useful in patients with statin-associated muscle adverse effects because it does not cause muscle pain or weakness. It can be used as monotherapy or as an add-on to other lipid-lowering therapy. Risks include hyperuricemia and tendon rupture.
Bile acid sequestrants (cholestyramine, colestipol, colesevelam) block intestinal bile acid reabsorption, forcing up-regulation of hepatic LDL receptors to recruit circulating cholesterol for bile synthesis. They have been shown to reduce cardiovascular mortality ((cholestyramine, colestipol, colesevelam) block intestinal bile acid reabsorption, forcing up-regulation of hepatic LDL receptors to recruit circulating cholesterol for bile synthesis. They have been shown to reduce cardiovascular mortality (14). Bile acid sequestrants are usually used with statins or with nicotinic acid to augment LDL-C reduction. They are the medications of choice for women who are or are planning to become pregnant. Statins are contraindicated in pregnancy because they maybe teratogenic due to the interruption of cholesterol synthesis, which is essential in fetal development. Bile acid sequestrants are safe, but their use is limited by adverse effects of bloating, nausea, cramping, and constipation. They may also increase triglycerides, so their use is contraindicated in patients with hypertriglyceridemia. Cholestyramine, colestipol, and colesevelam (but to a lesser degree), interfere with absorption of other medications—notably thiazides, beta-blockers, warfarin, digoxin, and thyroxine—an effect that can be decreased by administration at least 4 hours before or 1 hour after other medications. Bile acid sequestrants should be given with meals to increase their efficacy.(but to a lesser degree), interfere with absorption of other medications—notably thiazides, beta-blockers, warfarin, digoxin, and thyroxine—an effect that can be decreased by administration at least 4 hours before or 1 hour after other medications. Bile acid sequestrants should be given with meals to increase their efficacy.
The cholesterol absorption inhibitor ezetimibe inhibits intestinal absorption of cholesterol and phytosterol. ezetimibe inhibits intestinal absorption of cholesterol and phytosterol.Ezetimibe usually lowers LDL-C by 15 to 20% and causes small increases in HDL-C and a mild decrease in triglycerides (15). Ezetimibe can be used as monotherapy in patients intolerant to statins or added to statins for patients taking maximum statin doses with persistent LDL-C elevation. Adverse effects are infrequent.
PCSK9 monoclonal antibodies (alirocumab, evolocumab) are available as subcutaneous injections given once or twice per month. These drugs keep PCSK9 from attaching to LDL receptors, leading to improved function of these receptors. LDL-C is lowered by 40 to 70%. Cardiovascular outcomes trials with (alirocumab, evolocumab) are available as subcutaneous injections given once or twice per month. These drugs keep PCSK9 from attaching to LDL receptors, leading to improved function of these receptors. LDL-C is lowered by 40 to 70%. Cardiovascular outcomes trials withevolocumab and alirocumab showed a decrease in cardiovascular events in patients with prior atherosclerotic cardiovascular disease (9).
SiRNA targeting PCSK9 is given as a subcutaneous injection every 6 months. Inclisiran inhibits PCSK9 production in the liver, thereby prolonging activity of LDL receptors and lowering LDL-C levels. Although LDL-C is lowered, cardiovascular outcome trials with is given as a subcutaneous injection every 6 months. Inclisiran inhibits PCSK9 production in the liver, thereby prolonging activity of LDL receptors and lowering LDL-C levels. Although LDL-C is lowered, cardiovascular outcome trials withinclisiran are ongoing. Inclisiran can be used as an adjunct to diet and maximally tolerated statin therapy for patients with ASCVD or heterozygous familial hypercholesterolemia (16).
Dietary supplements that lower LDL-C levels include fiber supplements and other products containing plant sterols (sitosterol, campesterol) or stanols. Fiber supplements decrease cholesterol levels in multiple ways, including decreased absorption and increased excretion. Oat-based fiber supplements can decrease total cholesterol by up to 18%. Plant sterols and stanols decrease cholesterol absorption by displacing cholesterol from intestinal micelles and can reduce LDL-C by up to 10% without affecting HDL-C or triglycerides.
Cholesteryl ester transfer protein (CETP) inhibitors are a class of medications that have been found to lower LDL-C. CETP is a plasma glycoprotein produced in the liver and adipose tissue that circulates in the blood bound primarily to HDL, which mediates the transfer of cholesteryl esters from HDL to Apo B-containing particles (17).
Medications for homozygous familial hypercholesterolemia
Medications for homozygous familial hypercholesterolemia include statins, PCSK9 inhibitors, and evinacumab. Medications for homozygous familial hypercholesterolemia include statins, PCSK9 inhibitors, and evinacumab.Evinacumab is a recombinant human monoclonal antibody that binds to and inhibits angiopoietin-like protein 3, an inhibitor of lipoprotein lipase and endothelial lipase. It can decrease LDL-C (by 47%), triglyceride, and HDL-C. Evinacumab is given by intravenous infusion once monthly. It can cause gout, influenza-like illness, and infusion reactions (18).
Lomitapide is an inhibitor of microsomal triglyceride transfer protein that interferes with the secretion of triglyceride-rich lipoproteins in the liver and intestine. Dose is begun low and gradually titrated up about every 2 weeks. Patients must follow a diet with less than 20% of calories from fat. Lomitapide is an inhibitor of microsomal triglyceride transfer protein that interferes with the secretion of triglyceride-rich lipoproteins in the liver and intestine. Dose is begun low and gradually titrated up about every 2 weeks. Patients must follow a diet with less than 20% of calories from fat.Lomitapide can cause gastrointestinal adverse effects (eg, diarrhea, increased hepatic fat, elevated liver enzymes).
Procedural approaches
Procedural approaches are reserved for patients with severe hyperlipidemia (LDL-C > 300 mg/dL [> 7.74 mmol/L]) and no vascular disease. LDL apheresis, in which LDL is removed by extracorporeal plasma exchange, may be done in patients with LDL-C > 200 mg/dL (> 5.16 mmol/L) and vascular disease that is refractory to conventional therapy, such as occurs with familial hypercholesterolemia. Other options include ileal bypass (to block reabsorption of bile acids) and liver transplantation (which transplants LDL receptors). LDL apheresis is the procedure of choice in most instances when maximally tolerated therapy fails to lower LDL-C adequately. Apheresis is also the usual therapy in patients with the homozygous form of familial hypercholesterolemia who have limited or no response to pharmacotherapy.
Elevated LDL-C in children
Childhood risk factors besides family history and diabetes include cigarette smoking, hypertension, low HDL-C (< 35 mg/dL [< 0.9 mmol/L]), obesity, and physical inactivity.
The American Heart Association recommends dietary treatment for children with LDL-C > 110 mg/dL (> 2.8 mmol/L) (19).
Pharmacotherapy is recommended for children > 8 years and with either of the following:
Poor response to dietary therapy, LDL-C ≥ 190 mg/dL (≥ 4.9 mmol/L), and no family history of premature cardiovascular disease
LDL-C ≥ 160 mg/dL (≥ 4.13 mmol/L) and a family history of premature cardiovascular disease or ≥ 2 risk factors for premature cardiovascular disease
Medications used in children include many of the statins. Children with familial hypercholesterolemia may require a second medication to achieve LDL-C reduction of at least 50%.
Elevated triglycerides
Although it is unclear whether elevated triglycerides independently contribute to cardiovascular disease, they are associated with multiple metabolic abnormalities that contribute to coronary artery disease (eg, diabetes, metabolic syndrome). Guidelines support reducing triglycerides > 500 mg/dL to reduce the risk of pancreatitis (4). No guidelines specifically address treatment of elevated triglycerides in children.
The overall treatment strategy is to first implement lifestyle changes, including exercise, weight loss, and avoidance of concentrated dietary sugar and alcohol. Intake of 2 to 4 servings/week of marine fish high in omega-3 fatty acids may be effective, but the amount of omega-3 fatty acids is often lower than needed; supplemental doses may be helpful. The REDUCE-IT trial showed 4 grams of icosapent ethyl significantly reduced major adverse cardiac events when added to statin therapy in high-risk patients with TG > 150 mg/dL (1.7 mmol/L); however, it is uncertain whether the benefit was due to TG lowering or increase in eicosapentaenoic acid (EPA) (is to first implement lifestyle changes, including exercise, weight loss, and avoidance of concentrated dietary sugar and alcohol. Intake of 2 to 4 servings/week of marine fish high in omega-3 fatty acids may be effective, but the amount of omega-3 fatty acids is often lower than needed; supplemental doses may be helpful. The REDUCE-IT trial showed 4 grams of icosapent ethyl significantly reduced major adverse cardiac events when added to statin therapy in high-risk patients with TG > 150 mg/dL (1.7 mmol/L); however, it is uncertain whether the benefit was due to TG lowering or increase in eicosapentaenoic acid (EPA) (20). The STRENGTH trial did not confirm that another form of omega-3, which were lower in EPA, decreased major adverse cardiac events in patients with hypertriglyceridemia concomitant with low HDL (21).
In patients with diabetes, glucose levels should be tightly controlled. If these measures are ineffective, lipid-lowering medications should be considered. Patients with very high triglyceride levels (> 1,000 mg/dL [> 11 mmol/L]) may need to begin pharmacotherapy at diagnosis to more quickly reduce the risk of acute pancreatitis.
Fibrates reduce triglycerides by about 50%. They appear to stimulate endothelial lipoprotein lipase (LPL), leading to increased fatty acid oxidation in the liver and muscle and decreased hepatic VLDL synthesis. They also increase HDL-C by up to 20%. Fibrates can cause gastrointestinal adverse effects, including dyspepsia, abdominal pain, and elevated liver enzymes. They uncommonly cause cholelithiasis. Fibrates may potentiate muscle toxicity when used with statins and potentiate the effects of warfarin.. Fibrates may potentiate muscle toxicity when used with statins and potentiate the effects of warfarin.
Statins can be used in patients with triglycerides < 500 mg/dL (< 5.65 mmol/L) if LDL-C elevations are also present; statins may reduce both LDL-C and triglycerides through reduction of VLDL. If only triglycerides are elevated, fibrates are the medication of choice.
Omega-3 fatty acids in high doses (1 to 6 g a day of eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) can be effective in reducing triglycerides. The omega-3 fatty acids EPA and DHA are the active ingredients in marine fish oil or omega-3 capsules. Adverse effects include eructation and diarrhea. These effects may be decreased by giving the in high doses (1 to 6 g a day of eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) can be effective in reducing triglycerides. The omega-3 fatty acids EPA and DHA are the active ingredients in marine fish oil or omega-3 capsules. Adverse effects include eructation and diarrhea. These effects may be decreased by giving thefish oil capsules with meals in divided doses (eg, twice a day or 3 times a day). Omega-3 fatty acids can be a useful adjunct to other therapies. Prescription omega-3 fatty acid preparations are indicated for triglyceride levels > 500 mg/dL (> 5.65 mmol/L) (21).
The Apo CIII inhibitor (an antisense inhibitor of apo CIII) olezarsen lowers triglyceride levels in patients with familial chylomicronemic syndrome caused by lipoprotein lipase deficiency. It is given monthly ((an antisense inhibitor of apo CIII) olezarsen lowers triglyceride levels in patients with familial chylomicronemic syndrome caused by lipoprotein lipase deficiency. It is given monthly (22, 23).
Low HDL-C
Although higher HDL-C levels predict lower cardiovascular risk, it is not clear whether treatments to increase HDL-C levels decrease risk of death. Treatments for LDL-C and triglyceride reduction often increase HDL-C, and the 3 objectives can sometimes be achieved simultaneously.
No guidelines specifically address treatment of low HDL-C in children.
Pearls & Pitfalls
|
Treatment includes lifestyle changes such as an increase in exercise and weight loss. Alcohol raises HDL-C but is not recommended as a therapy because of its many other adverse effects. Medications may be successful in raising levels when lifestyle changes alone are insufficient, but it is uncertain whether raising HDL-C levels reduces mortality.
Nicotinic acid (niacin)Nicotinic acid (niacin) can increase HDL-C but is not routinely recommended due to its adverse effects. Its mechanism of action is unknown, but it appears to both increase HDL-C production and inhibit HDL-C clearance; it may also mobilize cholesterol from macrophages. Niacin also decreases triglycerides and, in doses of 1500 to 2000 mg a day, reduces LDL-C. However, neither the HPS2-THRIVE trial nor the AIM-HIGH trial showed any benefit to adding niacin to statin therapy in decreasing cardiac events in high-risk patients with known cardiovascular disease (24, 25).
Niacin causes flushing, pruritus, and nausea; premedication with low-dose aspirin may prevent these adverse effects. Extended-release preparations cause flushing less often. However, most over-the-counter slow-release preparations are not recommended. Niacin can cause liver enzyme elevations and occasionally liver failure, insulin resistance, and hyperuricemia and gout. It may also increase homocysteine levels. The combination of high doses of niacin with statins may increase the risk of myopathy.
Trials failed to show any benefit from fibrates when combined with statins (26, 27).
Elevated Lp(a)
The usual approach in patients with elevated Lp(a) is to lower LDL-C aggressively. LDL apheresis has been used to lower Lp(a) in patients with high Lp(a) levels and progressive vascular disease.
The upper limit of normal for Lp(a) is approximately 30 mg/dL (75 nmol/L), but values in African Americans tend to be higher. Few data exist to guide the treatment of elevated Lp(a) or to establish treatment efficacy. Niacin has been shown to decrease Lp(a); it can lower Lp(a) by > 20% at higher doses (The upper limit of normal for Lp(a) is approximately 30 mg/dL (75 nmol/L), but values in African Americans tend to be higher. Few data exist to guide the treatment of elevated Lp(a) or to establish treatment efficacy. Niacin has been shown to decrease Lp(a); it can lower Lp(a) by > 20% at higher doses (28). However, the clinical benefit of this lowering has not been born out in clinical trials (24, 25). There are several RNA-based therapies that lower Lp(a) levels in clinical development that show promise (29).
Treatment in patients with comorbidities
Treatment of diabetic dyslipidemia should always involve lifestyle changes and statins to reduce LDL-C. To decrease the risk of pancreatitis, fibrates can be used to decrease triglycerides when levels are > 500 mg/dL (> 5.65 mmol/L). Metformin lowers triglycerides, which may be a reason to choose it over other oral antihyperglycemic medications when treating diabetes. Some thiazolidinediones increase both HDL-C and LDL-C. Some thiazolidinediones also decrease triglycerides. These antihyperglycemic medications should not be chosen over lipid-lowering medications to treat lipid abnormalities in patients with diabetes but may be useful adjuncts. Patients with very high triglyceride levels and less than optimally controlled diabetes may have a better response to insulin than to oral antihyperglycemics. 5.65 mmol/L). Metformin lowers triglycerides, which may be a reason to choose it over other oral antihyperglycemic medications when treating diabetes. Some thiazolidinediones increase both HDL-C and LDL-C. Some thiazolidinediones also decrease triglycerides. These antihyperglycemic medications should not be chosen over lipid-lowering medications to treat lipid abnormalities in patients with diabetes but may be useful adjuncts. Patients with very high triglyceride levels and less than optimally controlled diabetes may have a better response to insulin than to oral antihyperglycemics.
There are increasing data to support use of glucagon-like peptide-1 receptor agonists (GLP-1RA) to improve glucose control, accelerate weight loss, improve cholesterol, and decrease cardiac risk in patients with diabetes (30, 31). Sodium-glucose transport protein 2 (SGLT2) inhibitors are controversial with regard to their effect on lipids, though they have been found to improve cardiac outcomes in diabetic patients (32, 33).
Treatment of dyslipidemia in patients with hypothyroidism, chronic kidney disease, liver disease, or a combination of these disorders involves treating the underlying disorders primarily and lipid abnormalities secondarily. Abnormal lipid levels in patients with low-normal thyroid function (high-normal TSH levels) improve with hormone replacement. Reducing the dosage of or stopping drugs that cause lipid abnormalities should be considered.
Monitoring treatment
Lipid levels should be monitored periodically after starting treatment. No data support specific monitoring intervals, but measuring lipid levels 2 to 3 months after starting or changing therapies and once or twice yearly after lipid levels are stabilized is common practice.
Liver and severe muscle toxicity with statin use occurs in 0.5 to 2% of all users. Routine monitoring of liver enzyme levels is not necessary, and routine measurement of creatine kinase (CK) is not useful to predict the onset of rhabdomyolysis. Muscle enzyme levels need not be checked regularly unless patients develop myalgias or other muscle symptoms. If statin-induced muscle damage is suspected, statin use is stopped and CK may be measured. When muscle symptoms subside, a lower dose or a different statin can be tried. If symptoms do not subside within 1 to 2 weeks of stopping the statin, another cause should be sought for the muscle symptoms (eg, polymyalgia rheumatica).
Treatment references
1. Daniels SR, Greer FR; Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008;122(1):198-208. doi:10.1542/peds.2008-1349
2. Harrison M, Moyna NM, Zderic TW, et al: Lipoprotein particle distribution and skeletal muscle lipoprotein lipase activity after acute exercise. Lipids Health Dis 11:64, 2012. doi:10.1186/1476-511X-11-64
3. American Heart Association: American Heart Association Recommendations for Physical Activity in Adults and Kids. Reviewed January 19, 2024. Accessed April 15, 2025
4. Grundy SM, Stone NJ, Bailey AL, et al: 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 139: e1082–e1143, 2019. doi: 10.1161/CIR.0000000000000625
5. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al: 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines [published correction appears in Circulation 2014 Jun 24;129(25 Suppl 2):S74-5]. Circulation 129(25 Suppl 2):S49–S73, 2014. doi:10.1161/01.cir.0000437741.48606.98
6. Khan SS, Coresh J, Pencina MJ, et al: Novel Prediction Equations for Absolute Risk Assessment of Total Cardiovascular Disease Incorporating Cardiovascular-Kidney-Metabolic Health: A Scientific Statement From the American Heart Association. Circulation 148(24):1982–2004, 2023. doi:10.1161/CIR.0000000000001191
7. Arnett DK, Blumenthal RS, Albert MA, et al: 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines [published correction appears in Circulation 2019 Sep 10;140(11):e647-e648. doi: 10.1161/CIR.0000000000000724.] [published correction appears in Circulation 2020 Jan 28;141(4):e59. doi: 10.1161/CIR.0000000000000754.] [published correction appears in Circulation 2020 Apr 21;141(16):e773. doi: 10.1161/CIR.0000000000000770.]. Circulation 140(11):e563–e595, 2019. doi:10.1161/CIR.0000000000000677
8. US Preventive Services Task Force, Mangione CM, Barry MJ, et al: Statin Use for the Primary Prevention of Cardiovascular Disease in Adults: US Preventive Services Task Force Recommendation Statement. JAMA 328(8):746-753, 2022. doi:10.1001/jama.2022.13044
9. Sabatine MS, Giugliano RP, Keech AC, et al: Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med 376:1713–1722, 2017.doi:10.1056/NEJMoa1615664
10. Schwartz GG, Steg PG, Szarek M, et al: Alirocumab and cardiovascular outcomes after acute coronary syndrome. New Engl J Med 379:2097–2107, 2018. doi: 10.1056/NEJMoa1801174.
11. Writing Committee, Lloyd-Jones DM, Morris PB, et al: 2022 ACC Expert Consensus Decision Pathway on the Role of Nonstatin Therapies for LDL-Cholesterol Lowering in the Management of Atherosclerotic Cardiovascular Disease Risk: A Report of the American College of Cardiology Solution Set Oversight Committee [published correction appears in J Am Coll Cardiol 2023 Jan 3;81(1):104]. J Am Coll Cardiol 80(14):1366-1418, 2022. doi:10.1016/j.jacc.2022.07.006
12. Goldberg AC, Leiter LA, Stroes ESG, et al: Effect of Bempedoic Acid vs Placebo Added to Maximally Tolerated Statins on Low-Density Lipoprotein Cholesterol in Patients at High Risk for Cardiovascular Disease: The CLEAR Wisdom Randomized Clinical Trial [published correction appears in JAMA. 2020 Jan 21;323(3):282]. JAMA 2019;322(18):1780-1788. doi:10.1001/jama.2019.16585
13. Ray KK, Bays HE, Catapano AL, et al: Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N Engl J Med 2019;380(11):1022-1032. doi:10.1056/NEJMoa1803917
14. The Lipid Research Clinics Coronary Primary Prevention Trial results: I. Reduction in incidence of coronary heart disease. JAMA 251(3):351–364, 1984. doi:10.1001/jama.1984.03340270029025
15. Cannon CP, Blazing MA, Giugliano RP, et al: Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med 372(25):2387–2397, 2015. doi:10.1056/NEJMoa1410489
16. Ray KK, Troquay RPT, Visseren FLJ, et al: Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endocrinol 11(2):109–119, 2023. doi:10.1016/S2213-8587(22)00353-9
17. Nicholls SJ, Ditmarsch M, Kastelein JJ, et al: Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: a randomized phase 2 trial. Nat Med 28(8):1672–1678, 2022. doi:10.1038/s41591-022-01936-7
18. Raal FJ, Rosenson RS, Reeskamp LF, et al: Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med 383(8):711–720, 2020. doi:10.1056/NEJMoa2004215
19. de Ferranti SD, Steinberger J, Ameduri R, et al: Cardiovascular Risk Reduction in High-Risk Pediatric Patients: A Scientific Statement From the American Heart Association. Circulation 139(13):e603-e634, 2019. doi:10.1161/CIR.0000000000000618
20. Bhatt DL, Steg PG, Miller M, et al: Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N Engl J Med 380(1):11–22, 2019. doi:10.1056/NEJMoa1812792
21. Nicholls SJ, Lincoff AM, Garcia M, et al. Effect of High-Dose Omega-3 Fatty Acids vs Corn Oil on Major Adverse Cardiovascular Events in Patients at High Cardiovascular Risk: The STRENGTH Randomized Clinical Trial. JAMA 2020;324(22):2268-2280. doi:10.1001/jama.2020.22258
22. Bergmark BA, Marston NA, Prohaska TA, et al: Olezarsen for Hypertriglyceridemia in Patients at High Cardiovascular Risk. N Engl J Med 390(19):1770–1780, 2024. doi:10.1056/NEJMoa2402309
23. Kamrul-Hasan ABM, Dutta D, Nagendra L, Mondal S, Bhattacharya S, Kalra S: Safety and Efficacy of the Novel RNA Interference Therapies for Hypertriglyceridemia and Mixed Hyperlipidemia Management: A Systematic Review and Meta-analysis. Endocr Pract 30(11):1103–1112, 2024. doi:10.1016/j.eprac.2024.08.013
24. AIM-HIGH Investigators, Boden WE, Probstfield JL, et al: Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy [published correction appears in N Engl J Med 367(2):189, 2012]. N Engl J Med 365(24):2255–2267, 2011. doi:10.1056/NEJMoa1107579
25. HPS2-THRIVE Collaborative Group, Landray MJ, Haynes R, et al: Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med 371(3):203–212, 2014. doi:10.1056/NEJMoa1300955
26. Das Pradhan A, Glynn RJ, Fruchart JC, et al: Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N Engl J Med 387(21):1923–1934, 2022. doi:10.1056/NEJMoa2210645
27. Ginsberg HN: The ACCORD (Action to Control Cardiovascular Risk in Diabetes) Lipid trial: what we learn from subgroup analyses. Diabetes Care 34 Suppl 2(Suppl 2):S107–S108, 2011. doi:10.2337/dc11-s203
28. Croyal M, Ouguerram K, Passard M, et al: Effects of extended-release nicotinic acid on apolipoprotein (a) kinetics in hypertriglyceridemic patients. Arterioscler Thromb Vasc Biol 35 (9): 2042–2047, 2015. doi:10.1161/ATVBAHA.115.305835
29. Yeang C, Karwatowska-Prokopczuk E, Su F, et al: Effect of Pelacarsen on Lipoprotein(a) Cholesterol and Corrected Low-Density Lipoprotein Cholesterol. J Am Coll Cardiol 79(11):1035–1046, 2022. doi:10.1016/j.jacc.2021.12.032
30. Hernandez AF, Green JB, Janmohamed S, et al: Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet 392(10157):1519–1529, 2018. doi:10.1016/S0140-6736(18)32261-X
31. Yao H, Zhang A, Li D, et al: Comparative effectiveness of GLP-1 receptor agonists on glycaemic control, body weight, and lipid profile for type 2 diabetes: systematic review and network meta-analysis. BMJ 384:e076410, 2024. doi:10.1136/bmj-2023-076410
32. Sánchez-García A, Simental-Mendía M, Millán-Alanís JM, Simental-Mendía LE: Effect of sodium-glucose co-transporter 2 inhibitors on lipid profile: A systematic review and meta-analysis of 48 randomized controlled trials. Pharmacol Res 160:105068, 2020. doi:10.1016/j.phrs.2020.105068
33. Wiviott SD, Raz I, Bonaca MP, et al: Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 380(4):347–357, 2019. doi:10.1056/NEJMoa1812389
Key Points
Elevated lipid levels are a risk factor for atherosclerosis and thus can lead to symptomatic coronary artery disease and peripheral arterial disease.
Causes of dyslipidemia include a sedentary lifestyle with excessive dietary intake of calories, saturated fat, cholesterol, and trans fats and/or genetic (familial) abnormalities of lipid metabolism.
Diagnose using serum lipid profile (measured total cholesterol, triglycerides, and high-density lipoprotein [HDL] cholesterol and calculated low-density lipoprotein [LDL] cholesterol and very low-density lipoprotein [VLDL] cholesterol).
Screening tests should be done at age 9 to 11 years and again at age 17 to 21 years (age 2 to 8 if there is a strong family history of severe hyperlipidemia or premature coronary artery disease or other risk factors); adults are screened every 5 years beginning at age 20.
Treatment with a statin is indicated to reduce risk of atherosclerotic cardiovascular disease for all patients in 4 major risk groups as defined by the American College of Cardiology/American Heart Association and for those without who have certain other combinations of risk factors and elevated lipid levels.
Optimize adherence, lifestyle changes, and statin usage before adding a non-statin drug; if a patient has an LDL-C level > 70 mg/dL (> 1.8 mmol/L) with high risk atherosclerotic cardiovascular disease, adding ezetimibe or PSCK9 inhibitor is reasonable.Optimize adherence, lifestyle changes, and statin usage before adding a non-statin drug; if a patient has an LDL-C level > 70 mg/dL (> 1.8 mmol/L) with high risk atherosclerotic cardiovascular disease, adding ezetimibe or PSCK9 inhibitor is reasonable.
Other treatment depends on the specific lipid abnormality but should always include lifestyle changes, treatment of hypertension and diabetes, smoking cessation, and in some patients with increased risk of myocardial infarction or death due to coronary artery disease, daily low-dose aspirin.Other treatment depends on the specific lipid abnormality but should always include lifestyle changes, treatment of hypertension and diabetes, smoking cessation, and in some patients with increased risk of myocardial infarction or death due to coronary artery disease, daily low-dose aspirin.
Drugs Mentioned In This Article





