



Ischemic stroke is sudden neurologic deficits that result from focal cerebral ischemia associated with permanent brain infarction (eg, positive results on diffusion-weighted MRI). Common causes are atherothrombotic occlusion of large arteries; cerebral embolism (embolic infarction); nonthrombotic occlusion of small, deep cerebral arteries (lacunar infarction); and proximal arterial stenosis with hypotension that decreases cerebral blood flow in arterial watershed zones (hemodynamic stroke). No cause is identified in one-third of ischemic strokes at the time of patient discharge; these strokes are categorized as cryptogenic. Diagnosis is clinical, but CT or MRI is performed to exclude hemorrhage and confirm the presence and extent of stroke. Acute thrombolytic therapy may be useful in certain patients. Depending on the cause of stroke, carotid endarterectomy or stenting, antiplatelet medications, or anticoagulants may help reduce risk of subsequent strokes.
Etiology of Ischemic Stroke
The following are the modifiable risk factors that contribute the most to increased risk of ischemic stroke:
Cigarette smoking
Insulin resistance
Abdominal obesity
Lack of physical activity
High-risk diet (eg, high in saturated fats, trans fats, and calories)
Psychosocial stress (eg, depression)
Heart disorders (particularly disorders that predispose to emboli, such as acute myocardial infarction, infective endocarditis, valvular heart disease, and atrial fibrillation)
Carotid artery stenosis
Use of certain drugs (eg, cocaine, amphetamines)
Use of exogenous estrogen
Unmodifiable risk factors include the following:
Prior stroke
Sex
Race/ethnicity
Older age
Family history of stroke
The most common causes of ischemic stroke can be classified as:
Cryptogenic (ie, no clear cardioembolic, lacunar, or atherosclerotic source; the most common classification)
Cardioembolism
Lacunar infarcts
Large-vessel atherosclerosis (the fourth most common cause)
Cryptogenic stroke
Stroke is classified as cryptogenic when one of the following occurs:
The diagnostic evaluation is incomplete.
No cause is identified despite an extensive evaluation (CT or MRI, cerebral vessel imaging, and cardiac evaluation).
There is more than one probable cause (eg, atrial fibrillation and ipsilateral carotid stenosis).
Embolic stroke of undetermined source (ESUS), a subcategory of cryptogenic stroke, is diagnosed when no source has been identified after sufficient diagnostic evaluation has excluded lacunar stroke, major cardioembolic sources, and ipsilateral steno-occlusive vessel disease (> 50% occlusion). Recent evidence suggests that symptomatic nonstenotic carotid disease with < 50% occlusion may be an important a cause of stroke (1).
Cardioembolism
Emboli may lodge anywhere in the cerebral arterial tree.
Emboli may originate as cardiac thrombi, especially in the following conditions:
Atrial fibrillation
Rheumatic heart disease (usually mitral stenosis)
Post-myocardial infarction
Vegetations on heart valves in bacterial or marantic endocarditis
Prosthetic heart valves
Mechanical circulatory assist devices (eg, left ventricular assist device, or LVAD [2])
Patent foramen ovale
Patent foramen ovale (PFO) is a frequent cause of cryptogenic stroke in younger patients, mainly caused by paradoxical embolism. Large PFO size and associated atrial septal aneurysm confer higher stroke risk. PFO closure may reduce recurrent stroke risk in carefully selected patients (3).
Other sources include clots that form after open-heart surgery and atheromas in neck arteries or in the aortic arch. Rarely, emboli consist of fat (from fractured long bones), air (in decompression sickness), or venous clots that pass from the right to the left side of the heart through a patent foramen ovale with shunt (paradoxical emboli). Emboli may dislodge spontaneously or after invasive cardiovascular procedures (eg, catheterization). Rarely, thrombosis of the subclavian artery results in embolic stroke in the vertebral artery or its branches.
Lacunar infarcts
Ischemic stroke can also result from lacunar infarcts. Lacunar infarcts are small strokes (≤ 1.5 cm) that occur in deep brain structures such as the basal ganglia, thalamus, and pons. They are most commonly due to underlying risk factors for cerebrovascular disease such as hypertension and diabetes. The small infarcts result from nonatherothrombotic obstruction of small, perforating arteries that supply deep cortical structures; the usual cause is lipohyalinosis (degeneration of the media of small arteries and replacement by lipids and collagen). Emboli may also cause lacunar infarcts.
When a stroke occurs that affects these locations in a patient who does not have typical vascular risk factors, or if the infarct is > 1.5 cm, an alternative mechanism, such as a central (cardiac or proximal arterial) embolic source, should be considered.
Large-vessel atherosclerosis
Large-vessel atherosclerosis can affect intracranial or extracranial arteries.
Atheromas, particularly if ulcerated, predispose to thrombi. Atheromas can occur in any major cerebral artery and are common at areas of turbulent flow, particularly at the carotid bifurcation. Partial or complete thrombotic occlusion occurs most often at the main trunk of the middle cerebral artery and its branches but is also common in the large arteries at the base of the brain, in deep perforating arteries, and in small cortical branches. The basilar artery and the segment of the internal carotid artery between the cavernous sinus and supraclinoid process are often occluded.
Other causes
Less common causes of ischemic stroke include vascular inflammation secondary to disorders such as acute or chronic meningitis, vasculitic disorders, and syphilis; dissection of intracranial arteries or the aorta; hypercoagulability disorders (eg, antiphospholipid syndrome, hyperhomocysteinemia, underlying malignancy); hyperviscosity disorders (eg, polycythemia, thrombocytosis, hemoglobinopathies, plasma cell disorders); and rare disorders (eg, fibromuscular dysplasia, Moyamoya disease, Binswanger disease).
In children, sickle cell disease is a common cause of ischemic stroke.
Any factor that impairs systemic perfusion (eg, carbon monoxide toxicity, severe anemia or hypoxia, polycythemia, hypotension) increases risk of all types of ischemic strokes. A stroke may occur along the borders between territories of arteries (watershed areas); in such areas, blood supply is normally low, particularly if patients have hypotension and/or if major cerebral arteries are stenotic.
Less commonly, ischemic stroke results from vasospasm (eg, during migraine, after subarachnoid hemorrhage, after use of sympathomimetic drugs such as cocaine or amphetamines) or venous sinus thrombosis (eg, during intracranial infection, postoperatively, peripartum, secondary to a hypercoagulability disorder).
Etiology references
1. Ospel JM, Kappelhof M, Ganesh A, Kallmes DF, Brinjikji W, Goyal M. Symptomatic non-stenotic carotid disease: current challenges and opportunities for diagnosis and treatment. J Neurointerv Surg. 2024;16(4):418-424. Published 2024 Mar 14. doi:10.1136/jnis-2022-020005
2. Caprio FZ, Sorond FA. Cerebrovascular Disease: Primary and Secondary Stroke Prevention. Med Clin North Am. 2019;103(2):295-308. doi:10.1016/j.mcna.2018.10.001
3. Caso V, Turc G, Abdul-Rahim AH, et al. European Stroke Organisation (ESO) Guidelines on the diagnosis and management of patent foramen ovale (PFO) after stroke. Eur Stroke J. 2024;9(4):800-834. doi:10.1177/23969873241247978
Pathophysiology of Ischemic Stroke
Inadequate blood flow in a single brain artery can often be compensated for by an efficient collateral system, particularly between the carotid and vertebral arteries via anastomoses at the circle of Willis and, to a lesser extent, between major arteries supplying the cerebral hemispheres. However, normal variations in the circle of Willis and in the caliber of various collateral vessels, atherosclerosis, and other acquired arterial lesions can interfere with collateral flow, increasing the chance that blockage of one artery will cause brain ischemia.
Some neurons die when perfusion is < 5% of normal for > 5 minutes; however, the extent of damage depends on the severity of ischemia. If it is mild, damage proceeds slowly; thus, even if perfusion is 40% of normal, 3 to 6 hours may elapse before brain tissue is completely lost. However, if severe ischemia persists > 15 to 30 minutes, all of the affected tissue dies (infarction). Damage occurs more rapidly during hyperthermia and more slowly during hypothermia. If tissues are ischemic but not yet irreversibly damaged, promptly restoring blood flow may reduce or reverse injury. For example, intervention may be able to salvage the moderately ischemic areas (penumbras) that often surround areas of severe ischemia; penumbras exist because of collateral flow.
Mechanisms of ischemic injury include:
Edema
Microvascular thrombosis
Programmed cell death (apoptosis)
Infarction with cell necrosis
Inflammatory mediators (eg, interleukin1-beta, tumor necrosis factor-alpha) contribute to edema and microvascular thrombosis. Edema, if severe or extensive, can increase intracranial pressure.
Many factors may contribute to necrotic cell death; they include loss of adenosine triphosphate (ATP) stores, loss of ionic homeostasis (including intracellular calcium accumulation), lipid peroxidative damage to cell membranes by free radicals (an iron-mediated process), excitatory neurotoxins (eg, glutamate), and intracellular acidosis due to accumulation of lactate.
Symptoms and Signs of Ischemic Stroke
Symptoms and signs of ischemic stroke depend on the part of brain affected. Patterns of neurologic deficits often suggest the affected artery (see table ), but correlation is often inexact.
Selected Stroke Syndromes
Symptoms and Signs | Syndrome |
|---|---|
Contralateral hemiparesis (maximal in the leg), urinary incontinence, apathy, confusion, poor judgment, mutism, grasp reflex, gait apraxia | Anterior cerebral artery (uncommon) |
Contralateral hemiparesis (worse in the arm and face than in the leg), dysarthria, hemianesthesia, contralateral homonymous hemianopia, aphasia (if the dominant hemisphere is affected) or apraxia and sensory neglect (if the nondominant hemisphere is affected) | Middle cerebral artery (common) |
Contralateral homonymous hemianopia, unilateral cortical blindness, memory loss, unilateral 3rd cranial nerve palsy, hemiballismus | Posterior cerebral artery |
Monocular loss of vision (amaurosis) | Ophthalmic artery (a branch of the internal carotid artery) |
Unilateral or bilateral cranial nerve deficits (eg, nystagmus, vertigo, dysphagia, dysarthria, diplopia, blindness), truncal or limb ataxia, spastic paresis, crossed sensory and motor deficits*, impaired consciousness, coma, death (if basilar artery occlusion is complete), tachycardia, labile blood pressure | Vertebrobasilar system |
Absence of cortical deficits plus one of the following:
| Lacunar infarcts |
* Ipsilateral facial sensory loss or motor weakness with contralateral body hemianesthesia or hemiparesis indicates a lesion at the pons or medulla. | |
Deficits may become maximal within several minutes of onset, typically in embolic stroke. Less often, deficits evolve slowly, usually over 24 to 48 hours (called evolving stroke or stroke in evolution), typically in atherothrombotic stroke.
In most evolving strokes, unilateral neurologic dysfunction (often beginning in one arm, then spreading ipsilaterally) extends without causing headache, pain, or fever. Progression is usually stepwise, interrupted by periods of stability.
A stroke is considered submaximal when, after it is complete, there is residual function in the affected area, suggesting viable tissue at risk of damage.
Embolic strokes often occur during the day; headache may precede neurologic deficits. Thrombi tend to occur during the night and thus thrombotic strokes are first noticed on awakening.
Lacunar infarcts may produce one of the classic lacunar syndromes (eg, pure motor hemiparesis, pure sensory hemianesthesia, combined hemiparesis and hemianesthesia, ataxic hemiparesis, dysarthria–clumsy hand syndrome); signs of cortical dysfunction (eg, aphasia) are absent. Multiple lacunar infarcts may result in multi-infarct dementia.
A seizure may occur at stroke onset, more often with embolic than thrombotic stroke. Seizures may also occur months to years later; late seizures result from scarring or hemosiderin deposition at the site of ischemia.
Occasionally, fever develops.
Deterioration during the first 48 to 72 hours after onset of symptoms, particularly progressively impaired consciousness, results more often from cerebral edema than from extension of the infarct. Unless the infarct is large or extensive, function commonly improves within the first few days; further improvement occurs gradually for up to 1 year.
Diagnosis of Ischemic Stroke
History and physical examination
Neuroimaging and bedside glucose testing
Evaluation to identify the cause
Diagnosis of ischemic stroke is suggested by sudden neurologic deficits referable to a specific arterial territory. Ischemic stroke must be distinguished from other causes of similar focal deficits (sometimes called stroke mimics, which are non-cerebrovascular disorders that cause focal neurologic signs. Conditions that can mimic strokes include:
Seizures (eg, with postictal paralysis)
CNS infections
Functional neurologic disorders (generally a diagnosis of exclusion)
Hypoglycemia
Headache, coma or stupor, and vomiting are more likely with hemorrhagic stroke than with an ischemic stroke.
When stroke is suspected, clinicians may use standardized criteria to grade severity and follow changes over time. This approach can be particularly useful as an outcome measure in efficacy studies. The National Institutes of Health Stroke Scale (NIHSS) is a 15-item scale to evaluate the patient's level of consciousness and language function and to identify motor and sensory deficits by asking the patient to answer questions and to perform physical and mental tasks. It is also useful for choosing appropriate treatment and predicting outcome.
The National Institutes of Health Stroke Scale*
Criterion | Finding | Score |
|---|---|---|
Level of consciousness (LOC) | Alert, keenly responsive | 0 |
Not alert, but can be aroused using minor stimulation to obey, answer, or respond | 1 | |
Not alert; requires repeated stimulation to attend or is obtunded and requires strong or painful stimulation to make purposeful movements | 2 | |
Responds only with reflex motor or autonomic effects or is totally unresponsive, flaccid, and areflexic | 3 | |
LOC questions† | Answers both correctly | 0 |
Answers one correctly | 1 | |
Answers both incorrectly | 2 | |
LOC commands‡ | Obeys both correctly | 0 |
Obeys one correctly | 1 | |
Obeys neither correctly | 2 | |
Gaze | Normal | 0 |
Partial gaze palsy; abnormal gaze in one or both eyes but no forced (involuntary) deviation and no total gaze paresis | 1 | |
Forced deviation or total gaze palsy (that is not corrected by the oculocephalic maneuver) | 2 | |
Visual field | No visual loss | 0 |
Partial hemianopia | 1 | |
Complete hemianopia | 2 | |
Bilateral hemianopia | 3 | |
Facial palsy | None | 0 |
Minor facial weakness | 1 | |
Partial facial weakness | 2 | |
Complete unilateral or bilateral paralysis | 3 | |
Motor arm function (score for both left and right sides) | No drift | 0 |
Drift before 10 seconds but does not hit bed or other support | 1 | |
Some effort against gravity | 2 | |
No effort against gravity | 3 | |
No movement | 4 | |
Motor leg function (score for both left and right sides) | No drift | 0 |
Drift before 5 seconds but does not hit bed or other support | 1 | |
Falls and hits bed before 5 seconds but has some effort against gravity | 2 | |
No effort against gravity | 3 | |
No movement | 4 | |
Limb ataxia | No ataxia | 0 |
Ataxia in 1 limb | 1 | |
Ataxia in 2 limbs | 2 | |
Untestable | — | |
Sensory | Normal | 0 |
Mild sensory loss | 1 | |
Severe sensory loss | 2 | |
Best language function§ | No aphasia | 0 |
Mild to moderate aphasia | 1 | |
Severe aphasia | 2 | |
Mute, global aphasia | 3 | |
Articulation/dysarthria | Normal articulation | 0 |
Mild to moderate dysarthria | 1 | |
Severe dysarthria (unintelligible or worse) | 2 | |
Untestable | — | |
Extinction or inattention | Absent | 0 |
Visual, tactile, auditory, spatial, or personal inattention or extinction to bilateral simultaneous stimulation in one of the sensory modalities | 1 | |
Profound hemi-inattention or extinction to more than one modality | 2 | |
* Total score is the sum of the scores for individual items. Degree of severity is indicated by the total score as follows:
| ||
† Patients are asked their age and the current month. | ||
‡ Patients are asked to open and close the eyes and to make a fist. | ||
§ For aphasia:
| ||
Evaluation of ischemic stroke requires assessment of the brain parenchyma, vascular system (including the heart and large arteries), and blood.
Differentiating clinically between the types of stroke is imprecise; however, some clues based on symptom progression, time of onset, and type of deficit can help.
Although diagnosis is clinical, neuroimaging and bedside glucose testing are mandatory.
Distinction between lacunar, embolic, and thrombotic stroke based on history, examination, and neuroimaging is not always reliable, so tests to identify common or treatable causes and risk factors for all types of strokes are routinely performed. The evaluation should assess the following systems:
Cardiac (eg, atrial fibrillation, potential structural sources of emboli)
Vascular (eg, critical arterial stenosis detected by vascular imaging)
Blood (eg, diabetes, dyslipidemia, hypercoagulability)
Strokes are categorized as cryptogenic when a cause cannot be identified.
Brain assessment
Neuroimaging with CT or MRI is performed first to exclude intracerebral hemorrhage, subdural or epidural hematoma, and a rapidly growing, bleeding, or suddenly symptomatic tumor. CT evidence of even a large anterior circulation ischemic stroke may be subtle during the first few hours; changes may include effacement of sulci or the insular cortical ribbon, loss of the gray-white junction between cortex and white matter, and a dense middle cerebral artery sign. Within 6 to 12 hours of ischemia, medium-sized to large infarcts start to become visible as hypodensities; small infarcts (eg, lacunar infarcts) may be visible only with MRI.
Diffusion-weighted MRI (highly sensitive for early ischemia) can be performed immediately after initial CT neuroimaging.
Imaging of the cervical and intracranial vessels is generally recommended as a part of the routine workup to determine etiology and guide further management. Guidelines recommend that all patients with clinical suspicion of large vessel occlusion should undergo vessel imaging (CTA or MRA), especially within 24 hours of symptom onset given advances in acute stroke treatment.

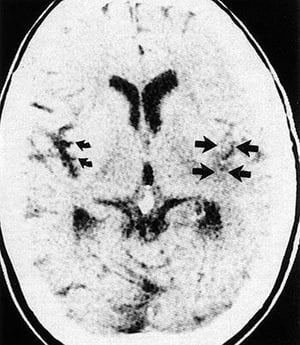
This CT scan shows effacement of the sylvian fissure and insular ribbon (straight arrows) on the infarcted side of brain compared with the normal insular ribbon (curved arrows).
This CT scan shows effacement of the sylvian fissure and insular ribbon (straight arrows) on the infarcted side of brai
By permission of the publisher. From Geremia G, Greenlee W. In Atlas of Cerebrovascular Disease. Edited by PB Gorelick and MA Sloan. Philadelphia, Current Medicine, 1996.

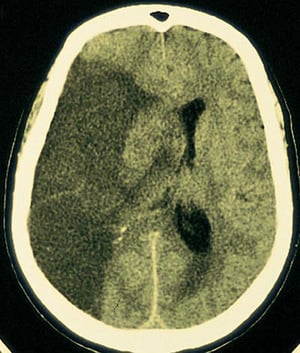
A large lucent infarction is seen in the area of brain supplied by the right middle cerebral artery.
A large lucent infarction is seen in the area of brain supplied by the right middle cerebral artery.
By permission of the publisher. From Furie K, et al: Cerebrovascular disease. In Atlas of Clinical Neurology. Edited by RN Rosenberg. Philadelphia, Current Medicine, 2002.

This noncontrast head CT scan shows a hyperdense left middle cerebral artery. This finding indicates a focal clot in the left middle cerebral artery (arrow).
This noncontrast head CT scan shows a hyperdense left middle cerebral artery. This finding indicates a focal clot in th
Image courtesy of Ji Y. Chong, MD.

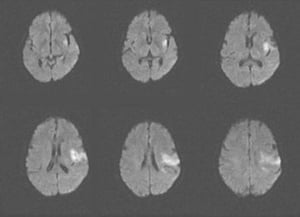
This MRI scan shows an area of restricted diffusion consistent with an acute ischemic stroke in the left insular and frontal lobes.
This MRI scan shows an area of restricted diffusion consistent with an acute ischemic stroke in the left insular and fr
Image courtesy of Ji Y. Chong, MD.

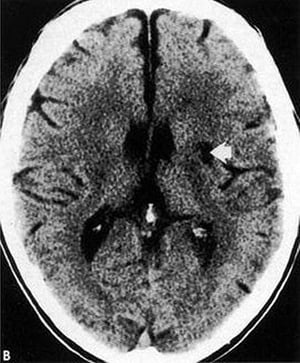
This CT scan shows a low-density, well-defined lacunar infarct (arrow) within the basal ganglia.
This CT scan shows a low-density, well-defined lacunar infarct (arrow) within the basal ganglia.
By permission of the publisher. From Geremia G, Greenlee W. In Atlas of Cerebrovascular Disease. Edited by PB Gorelick and MA Sloan. Philadelphia, Current Medicine, 1996.
This CT scan shows effacement of the sylvian fissure and insular ribbon (straight arrows) on the infarcted side of brain compared with the normal insular ribbon (curved arrows).
This CT scan shows effacement of the sylvian fissure and insular ribbon (straight arrows) on the infarcted side of brai
By permission of the publisher. From Geremia G, Greenlee W. In Atlas of Cerebrovascular Disease. Edited by PB Gorelick and MA Sloan. Philadelphia, Current Medicine, 1996.
A large lucent infarction is seen in the area of brain supplied by the right middle cerebral artery.
A large lucent infarction is seen in the area of brain supplied by the right middle cerebral artery.
By permission of the publisher. From Furie K, et al: Cerebrovascular disease. In Atlas of Clinical Neurology. Edited by RN Rosenberg. Philadelphia, Current Medicine, 2002.
This noncontrast head CT scan shows a hyperdense left middle cerebral artery. This finding indicates a focal clot in the left middle cerebral artery (arrow).
This noncontrast head CT scan shows a hyperdense left middle cerebral artery. This finding indicates a focal clot in th
Image courtesy of Ji Y. Chong, MD.
This MRI scan shows an area of restricted diffusion consistent with an acute ischemic stroke in the left insular and frontal lobes.
This MRI scan shows an area of restricted diffusion consistent with an acute ischemic stroke in the left insular and fr
Image courtesy of Ji Y. Chong, MD.
This CT scan shows a low-density, well-defined lacunar infarct (arrow) within the basal ganglia.
This CT scan shows a low-density, well-defined lacunar infarct (arrow) within the basal ganglia.
By permission of the publisher. From Geremia G, Greenlee W. In Atlas of Cerebrovascular Disease. Edited by PB Gorelick and MA Sloan. Philadelphia, Current Medicine, 1996.
Cardiac causes
Testing for cardiac causes typically includes ECG, telemetry or Holter monitoring, serum troponin, and transthoracic or transesophageal echocardiography. Transthoracic or transesophageal echocardiogram are useful to detect intracardiac thrombi, valvular disease, and patent foramen ovale (PFO). PFO is one of the frequent causes of embolic strokes.
Implantable cardiac monitors are useful for detecting underlying atrial arrhythmias in patients with cryptogenic stroke (1).
Vascular causes
For vascular causes, testing may include magnetic resonance angiography (MRA), CT angiography (CTA), carotid and transcranial duplex ultrasound, and conventional angiography. The choice and sequence of testing is individualized, based on clinical findings. MRA, CTA, and carotid ultrasound all show the anterior circulation; however, MRA and CTA provide better images of the posterior circulation than carotid ultrasound. In general, CTA is preferred to MRA because motion artifacts are avoided. Usually, CTA or MRA should be performed urgently but should not delay treatment with IV tPA if it is indicated.
Blood-related causes
Routine blood testing typically includes complete blood count (CBC), metabolic panel, prothrombin time/partial thromboplastin time (PT/PTT), fasting blood glucose, hemoglobin A1C, and lipid profile.
Depending on which causes are clinically suspected, additional tests may include measurement of homocysteine, testing for thrombotic disorders (antiphospholipid antibodies, protein S, protein C, antithrombin III, factor V Leiden), testing for rheumatic disorders (eg, antinuclear antibodies, rheumatoid factor, erythrocyte sedimentation rate), syphilis serologic testing, hemoglobin electrophoresis, and a urine drug screen for cocaine and amphetamines.
Diagnosis reference
1. Sanna T , Diener H-C., Passman RS, et al. Cryptogenic stroke and underlying atrial fibrillation. N Engl J Med. 370:2478–2486, 2014. doi: 10.1056/NEJMoa1313600
Treatment of Ischemic Stroke
General stroke treatments
Acute antihypertensive therapy only in certain circumstances
For acute treatment, sometimes reperfusion with recombinant tissue plasminogen activator or tenecteplase and/or mechanical thrombectomy
Sometimes carotid endarterectomy or stenting
Antiplatelet therapy
Sometimes anticoagulation
Long-term control of risk factors
For long-term treatment, rehabilitation
Acute stroke treatment
Guidelines for early management of stroke are available. See Guidelines for the Early Management of Patients With Acute Ischemic Stroke (2019) from the American Heart Association/American Stroke Association. Patients with acute ischemic strokes are usually hospitalized.
Supportive measures such as the following may be needed during initial evaluation and stabilization.
Airway support and ventilatory assistance if decreased consciousness or bulbar dysfunction compromises the airway
Supplemental oxygen only if needed to maintain oxygen saturation > 94%
Correction of hyperthermia (temperature > 38° C) with antipyretic medication and identifying and treating the cause of hyperthermia
Treatment of hypoglycemia (blood glucose < 60 mg/dL)
Treatment of hyperglycemia to lower blood glucose to 140 to 180 mg/dL while closely monitoring for hypoglycemia
Perfusion of an ischemic brain area may require an elevated blood pressure (BP) because autoregulation is lost; thus, BP should not be decreased except in the following cases:
Signs of other end-organ damage (eg, aortic dissection, acute myocardial infarction, pulmonary edema, hypertensive encephalopathy, retinal hemorrhages, acute renal failure).
Use of recombinant tissue plasminogen activator (tPA) and/or mechanical thrombectomy is likely.
If BP is ≥ 220 mm Hg systolic or ≥ 120 mm Hg diastolic on 2 successive readings 15 minutes apart, lowering BP by 15% in the 24 hours after stroke onset is reasonable.
For patients who are eligible for acute reperfusion therapy, intravenous treatment to achieve BP < 180/105 mm Hg before starting IV thrombolysis is initiated with one of the following:
Labetalol
Nicardipine
Clevidipine
Patients with presumed thrombi or emboli may be treated with one or a combination of the following:
Tissue plasminogen activator (tPA), thrombolysis-in-situ, and/or mechanical thrombectomy
Antiplatelet medications
Anticoagulants
Recombinant tPA (alteplase) can be used for patients with acute ischemic stroke up to 3 hours after symptom onset if they have no contraindications to tPA (see table ). Some experts recommend using tPA up to 4.5 hours after symptom onset (see Expansion of the Time Window for Treatment of Acute Ischemic Stroke With Intravenous Tissue Plasminogen Activator); however, between 3 hours and 4.5 hours after symptom onset, additional exclusion criteria apply (see table ). Thus, tPA must be given within 4.5 hours of symptom onset—a difficult requirement. Because the precise time of symptom onset may not be known, clinicians must start timing from the moment the patient was last observed to be well.
Although tPA can cause fatal or other symptomatic brain hemorrhage, patients treated with tPA strictly according to protocols have a higher likelihood of functional neurologic recovery. When tPA is given incorrectly (eg, when given despite the presence of exclusion criteria), risk of hemorrhage due to tPA is high mainly for patients who have had stroke; risk of brain hemorrhage is very low (approximately 0.5%; 95% confidence interval of 0 to 2.0% [1]) for patients who have had a stroke mimic (eg, hemiplegic migraine, certain CNS infections, postictal paralysis, functional neurologic disorders).
Only clinicians experienced in stroke management should use tPA to treat patients with acute stroke. Inexperienced physicians are more likely to violate protocols, resulting in more brain hemorrhages and deaths. If experienced clinicians are not available on site, consultation with an expert at a stroke center (including video evaluation of the patient [telemedicine]), if possible, may enable these clinicians to use tPA. Because most poor outcomes result from failure to strictly adhere to the protocol, a checklist of inclusion and exclusion criteria should be used.
Before treatment with tPA, the following are required:
Brain hemorrhage must be excluded by CT
Systolic BP must be < 185 mm Hg
Diastolic BP must be < 105 mm Hg
Blood glucose must be > 50 mg/dL
Antihypertensives (IV nicardipine, IV labetalol, IV clevidipine) may be given to maintain BP < 180/105 mm Hg for at least 24 hours after treatment with tPA.
Dose of tPA is 0.9 mg/kg IV (maximum dose 90 mg); 10% is given by rapid IV injection over 1 minute, and the remainder by constant infusion over 60 minutes. Vital signs are closely monitored for 24 hours after treatment. Any bleeding complications are aggressively managed. Anticoagulants and antiplatelet medications are not used within 24 hours of treatment with tPA.
Tenecteplase is a reasonable alternative to alteplase (2). Tenecteplase differs from alteplase by three amino acids in its glycoprotein structure. Although there is no significant difference in outcomes or adverse events, rates of reperfusion are higher in patients treated with tenecteplase. Compared with alteplase, tenecteplase has greater fibrin binding, increased conversion of plasminogen to plasmin, greater resistance to plasminogen activator inhibitor, and easier administration as a single bolus.
Patients who do not quality for IV thrombolysis should be given an antiplatelet medication (usually aspirin 325 mg orally) when they are admitted to the hospital. Contraindications to antiplatelet medications include aspirin-induced or nonsteroidal anti-inflammatory drug (NSAID)-induced asthma or urticaria, other hypersensitivity to aspirin or to tartrazine, acute gastrointestinal bleeding, glucose-6-phosphate dehydrogenase (G6PD) deficiency, and use of warfarin.
Exclusion Criteria for Use of Tissue Plasminogen Activator in Stroke
Exclusion criteria < 3 hours after symptom onset |
Intracranial hemorrhage on CT scan |
Multilobar infarct (hypodensity in more than one third of the territory supplied by the middle cerebral artery) on CT scan |
Presentation suggesting subarachnoid hemorrhage even if CT is negative |
History of intracranial hemorrhage |
Intra-axial intracranial tumor |
History of stroke or severe head trauma within the past 3 months |
Systolic BP > 185 mm Hg or diastolic BP > 110 mm Hg after antihypertensive treatment |
Intracranial or spinal surgery within the past 3 months |
Active internal bleeding |
Gastrointestinal cancer or bleeding within 21 days |
Suspected bleeding disorder |
Platelet count < 100,000/mcL |
Possibly arterial puncture of a noncompressible vessel in the 7 days before stroke onset |
Use of a treatment dose of low molecular weight heparin within previous 24 hours |
Coagulopathy, international normalized ratio (INR) > 1.7, or prothrombin time (PT) > 15 seconds, activated partial thromboplastin time (aPTT) > 40 seconds |
Use of a direct thrombin inhibitor or direct factor Xa inhibitor within 48 hours and with evidence of anticoagulant effect, detected by tests such as PTT, INR, and appropriate factor Xa activity assays |
Aortic arch dissection that is known or suspected to cause the ischemic stroke |
Bacterial endocarditis |
Relative exclusion criteria (2) |
Rapidly decreasing symptoms |
Major surgery or serious trauma in the past 14 days |
Urinary tract hemorrhage in the past 21 days |
Seizure at onset with postictal residual neurologic deficits |
Pregnancy |
Acute myocardial infarction in the past 3 months |
Additional considerations 3–4.5 hours after symptom onset* |
Age > 80 years |
Use of oral anticoagulants regardless of INR |
Baseline NIH stroke score > 25 |
A history of both stroke and diabetes mellitus |
* The benefit for patients with these characteristics is less clear. |
NIH = National Institutes of Health. Powers WJ, Rabinstein AA, Ackerson T, et al. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association [published correction appears in Stroke. 2019 Dec;50(12):e440-e441. doi: 10.1161/STR.0000000000000215.]. Stroke. 2019;50(12):e344-e418. doi:10.1161/STR.0000000000000211 |
Retrospective analyses have led to narrowing the list of absolute contraindications to using alteplase in the therapeutic window after onset of stroke symptoms (3, 4). Nevertheless, if patients have any of the following conditions, the risk of bleeding with tPA is increased and should be weighed against the anticipated benefits [5]):
Recent major surgery or procedure (eg, coronary artery bypass graft, obstetrical delivery, organ biopsy, previous puncture of noncompressible vessels)
Cerebrovascular disease
Recent intracranial hemorrhage
Recent gastrointestinal or genitourinary bleeding
Recent trauma
Hypertension (systolic BP > 175 mm Hg or diastolic BP > 110 mm Hg
Acute pericarditis
Subacute bacterial endocarditis
Hemostatic defects including those due to severe hepatic or renal disease
Significant hepatic dysfunction
Pregnancy
Hemorrhagic diabetic retinopathy or other hemorrhagic ophthalmic conditions
Septic thrombophlebitis or occluded arteriovenous cannula at an infected site
Advanced age (> 77 years)
Current use of anticoagulants (eg, warfarin)
Thrombolysis-in-situ (angiographically directed intra-arterial thrombolysis) of a thrombus or embolus can be considered when a clot is too distal to be accessed by catheters (eg, distal A2 [anterior cerebral artery distal to the anterior communicating artery]).
Mechanical thrombectomy (angiographically directed intra-arterial removal of a thrombus or embolus by a stent retriever device) is standard of care in large stroke centers for patients with recent large-vessel occlusion in the anterior circulation. Evidence for the efficacy of mechanical thrombectomy in patients with posterior circulation strokes is growing (6).
Mechanical thrombectomy had previously been restricted to use within 6 hours of symptom onset in patients with internal carotid artery or middle cerebral artery occlusion. However, later treatment may be justified at comprehensive stroke centers when clinical and/or imaging findings that suggest presence of a substantial amount of tissue at risk of infarction (penumbra). The volume of infarcted tissue and at-risk underperfused tissue (ischemic penumbra) can be identified using perfusion CT or perfusion MRI. A sizeable mismatch between the infarct and at-risk volume identified by diffusion-weighted or perfusion-weighted imaging suggests that substantial penumbra is still potentially salvageable. In the DEFUSE 3 trial, benefit was evident up to 16 hours after symptom onset in patients with a small infarct and a larger penumbra; both findings are based on imaging criteria (7). In the DAWN trial, benefit was evident up to 24 hours after symptom onset in patients with a large mismatch between infarct volume based on imaging and severity of the clinical deficit based on clinical criteria (8).
In the past, clinical trials restricted mechanical thrombectomy to patients with an NIHSS score > 6; however, recent evidence supports the usefulness of mechanical thrombectomy in patients with an NIHSS < 6 (9).
Evidence does not support using mechanical thrombectomy alone in patients who are eligible for intravenous thrombolysis. For patients with acute ischemic stroke due to emergent large vessel occlusion, administering intravenous thrombolysis before mechanical thrombectomy is recommended when criteria for intravenous thrombolysis are met, and intravenous thrombolysis should not be omitted in favor of mechanical thrombectomy alone (10). Devices used to remove thrombi are being improved, and recent models reestablish perfusion in 90 to 100% of patients.


This cerebral angiogram shows an occlusion of the proximal left middle cerebral artery (MCA) on anteroposterior and lateral views (1, 2). After mechanical thrombectomy, the angiogram shows restored blood flow in the left MCA on anteroposterior and lateral views (3, 4).
Image courtesy of Ji Y. Chong, MD.
Oral antiplatelet medications are used in acute stroke treatment to reduce the risk of recurrent disabling stroke. The following may be used:
Aspirin 81 to 325 mg within 48 hours of stroke onset
Dual antiplatelet therapy with aspirin plus clopidogrel within 24 hours of stroke onset for patients with minor stroke or at high risk of a transient ischemic attack (TIA; ABCD2 score ≥ 4)
Dual antiplatelet therapy with aspirin 81 mg and ticagrelor (possibly beneficial for patients with clopidogrel resistance CYP2C19 loss of function allele, especially if an extracranial or intracranial stent is used)
Aspirin given within 48 hours reduces the risk of early recurrent stroke and death (11).
If patients have had a TIA or minor stroke, clopidogrel plus aspirin given within 24 hours of symptom onset and continued for 21 days appears more effective than aspirin alone for reducing risk of stroke in the first 90 days and does not increase risk of hemorrhage (12). However, prolonged (eg, > 3 months) use of clopidogrel plus aspirin is avoided because it has no advantage over aspirin alone in long-term secondary stroke prevention and results in more bleeding complications.
Anticoagulation is usually avoided in the acute stage because risk of hemorrhage (hemorrhagic transformation) is higher, especially with large infarcts. However, anticoagulation with heparin or low molecular weight heparin is used for stroke caused by cerebral venous thrombosis and sometimes for stroke caused by cervical artery dissection. Anticoagulation can also be used in patients at high risk of recurrent cardiac emboli (eg, those with cardiac thrombi or mechanical valves).
Long-term stroke treatment
Supportive care is continued during convalescence:
Controlling hyperglycemia and fever can limit brain damage after stroke, leading to better functional outcomes.
Screening for dysphagia before patients begin eating, drinking, or receiving oral medications can help identify patients at increased risk of aspiration; it should be performed by a speech-language pathologist or other trained health care professional.
Enteral nutrition, if needed, should be started within 7 days of admission after an acute stoke.
Intermittent pneumatic compression (IPC) for deep venous thrombosis prophylaxis is recommended for immobile stroke patients without contraindications.
Low molecular weight heparin may be given to immobile stroke patients without contraindications.
Measures to prevent pressure ulcers are started early.
Physical therapy to help maximize function and prevent sarcopenia and joint contractures
Long-term management focuses on prevention of recurrent stroke (secondary prevention). Modifiable risk factors (eg, hypertension, diabetes, smoking, alcohol use disorder, dyslipidemia, obesity) are treated. Reducing systolic BP may be more effective when the target BP is < 120 mm Hg rather than the typical level (< 140 mm Hg) (13). The timeline for reducing BP to these levels should be determined based on each patient's health status and risk of recurrent stroke or other cardiovascular events.
Depression often occurs after a stroke and may interfere with recovery. Treatment of depression may aid in recovery, Clinicians should ask patients whether they are feeling sad or have lost interest or pleasure in doing formerly enjoyable activities. Clinicians should also ask family members whether they have noticed any signs of depression in the patient.
Extracranial carotid endarterectomy or stenting is indicated for patients with recent nondisabling, submaximal stroke attributed to an ipsilateral carotid obstruction of 70 to 99% of the arterial lumen or to an ulcerated plaque if life expectancy is at least 5 years. In other symptomatic patients (eg, patients with TIAs), endarterectomy or stenting combined with antiplatelet therapy is indicated for carotid obstruction of ≥ 60% with or without ulceration if life expectancy is at least 5 years. These procedures should be performed by surgeons and interventionists who have a successful record with the procedure (ie, morbidity and mortality rate of < 3%) in the hospital where it will be performed. For many patients, carotid stenting with an emboli-protection device (a type of filter) is preferred to endarterectomy, particularly if patients are ≥ 70 years and have a high surgical risk. Carotid endarterectomy and stenting are equally effective for stroke prevention. In the periprocedural period, myocardial infarction is more likely after endarterectomy, and recurrent stroke is more likely after stenting.
Extracranial vertebral angioplasty and/or stenting can be used in certain patients with recurrent symptoms of vertebrobasilar ischemia despite optimal medical treatment and a vertebral artery obstruction of 50 to 99%.
Intracranial major artery angioplasty and/or stenting may be effective in patients when optimal treatment with medications has been ineffective. Key factors to consider are patient characteristics (eg, control of risk factors, adherence to the medication regimen), timing of the procedure (> 3 weeks after the stroke), and the interventionist's experience. The rate of periprocedural adverse events can be acceptably low after percutaneous transluminal angioplasty and stenting when these factors are considered (14).
Endovascular closure of a patent foramen ovale plus use of antiplatelet therapy is recommended for patients < 60 years with an embolic stroke of undetermined cause despite extensive evaluation (4, 15).
Oral antiplatelet medications are used to prevent subsequent noncardioembolic (eg, atherothrombotic, lacunar, cryptogenic) strokes (secondary prevention). The following may be used:
Aspirin 81 or 325 mg once a day
Clopidogrel
Ticagrelor
A combination product of aspirin and extended-release dipyridamole
In patients taking oral anticoagulants, antiplatelet medications additively increase risk of bleeding and are thus usually avoided; however, aspirin is occasionally used simultaneously with warfarin in certain high-risk patients. Clopidogrel is indicated for patients who are allergic to aspirin. If ischemic stroke recurs or if a coronary artery stent becomes blocked while patients are taking clopidogrel, clinicians should suspect impaired metabolism of clopidogrel (ineffective conversion of clopidogrel to its active form because cytochrome P-450 2C19 [CYP2C19] activity is reduced); a test to determine CYP2C19 status (eg, genetic testing for CYP450 polymorphisms) is recommended. If impaired metabolism is confirmed, aspirin or combination aspirin and ticagrelor is a reasonable alternative.
Clopidogrel plus aspirin, if started during acute treatment, is given for only a short time (eg, < 3 weeks) because it has no advantage over aspirin alone in long-term secondary stroke prevention and results in more bleeding complications. Clopidogrel plus aspirin before and for ≥ 30 days after stenting is indicated, usually for ≤ 6 months; if patients cannot tolerate clopidogrel or have clopidogrel resistance due to CYP2C19 loss of allele, ticagrelor can be substituted.
Oral anticoagulants are indicated for secondary prevention of cardioembolic strokes (as well as primary prevention), but are contraindicated for thrombotic strokes. Adjusted-dose warfarin (a vitamin K antagonist) with a target international normalized ratio (INR) of 2 to 3 is used for certain patients with nonvalvular or valvular atrial fibrillation. A target INR of 2.5 to 3.5 is used if patients have a mechanical prosthetic cardiac valve. Efficacious alternatives to warfarin for patients with nonvalvular atrial fibrillation include the following direct oral anticoagulants:
Dabigatran (a direct thrombin inhibitor)
Apixaban (a direct factor Xa inhibitor)
Rivaroxaban (a direct factor Xa inhibitor)
Edoxaban (a direct factor Xa inhibitor)
The main advantage of these direct oral anticoagulants, compared to warfarin, is ease of use (eg, no need to check anticoagulation level with a blood test after the initial dose or to use a parenteral anticoagulant such as unfractionated heparin given by continuous infusion when transitioning from parenteral to oral anticoagulants). Their main disadvantage is cost. In case a hemorrhagic complication occurs, the antidote for dabigatran is idarucizumab (14), and the antidote for apixaban or rivaroxaban is andexanet alfa. Efficacy and safety of combining any of these new anticoagulants with an antiplatelet medication have not been established.
Statins are used to prevent recurrent strokes; lipid levels must be decreased by substantial amounts (16). Statin therapy (atorvastatin, rosuvastatin, simvastatin, pravastatin) is recommended for patients with evidence of atherosclerotic stroke and LDL (low-density lipoprotein) cholesterol ≥ 100 mg/dL. A reasonable LDL cholesterol target is a 50% reduction or a level of < 70 mg/dL.
Treatment references
1. Tsivgoulis G, Zand R, Katsanos AH, et al. Safety of intravenous thrombolysis in stroke mimics: prospective 5-year study and comprehensive meta-analysis. Stroke. 46 (5):1281–1287, 2015. doi: 10.1161/STROKEAHA.115.009012
2. Menon BK, Singh N, Sylaja, PN. Tenecteplase use in patients with acute ischaemic stroke. Lancet. 2023;401(10377):618-619. doi:10.1016/S0140-6736(22)02633-2
3. Frank B, Grotta JC,.Alexandrov AV, et al. Thrombolysis in stroke despite contraindications or warnings? Stroke. 2013;44(3):727-733. doi:10.1161/STROKEAHA.112.674622
4. Powers WJ, Rabinstein AA, Ackerson T, et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2019 Dec;50(12):e440-e441. doi: 10.1161/STR.0000000000000215.]. Stroke. 2019;50(12):e344-e418. doi:10.1161/STR.0000000000000211
5. Highlights of prescribing information for alteplase. Accessed April 28, 2025.
6. Jovin TG, Li C, Wu L, et al. Trial of thrombectomy 6 to 24 hours after stroke due to basilar-artery occlusion. N Engl J Med. 2022; 387:1373-1384, 2022. doi: 10.1056/NEJMoa2207576
7. Albers GW, Marks MP, Kemp S, et al. Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. N Engl J Med. 2018;378(8):708-718. doi:10.1056/NEJMoa1713973
8. Nogueira RG, Jadhav AP, Haussen DC, et al. Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct. N Engl J Med. 2018;378(1):11-21. doi:10.1056/NEJMoa1706442
9. Abecassis IJ, Almallouhi E, Chalhoub R, et al. Outcomes after endovascular mechanical thrombectomy for low compared to high National Institutes of Health Stroke Scale (NIHSS): A multicenter study. Clin Neurol Neurosurg. 2023;225:107592. doi:10.1016/j.clineuro.2023.107592
10. Masoud HE, de Havenon A, Castonguay AC, et al. 2022 Brief practice update on intravenous thrombolysis before thrombectomy in patients with large vessel occlusion acute ischemic stroke: A statement from Society of Vascular and Interventional Neurology Guidelines and Practice Standards (GAPS) Committee. Stroke. Vasc Interv Neurol. 2 (4) 2022. doi: 10.1161/SVIN.121.000276
11. CAST: randomised placebo-controlled trial of early aspirin use in 20,000 patients with acute ischaemic stroke. CAST (Chinese Acute Stroke Trial) Collaborative Group. Lancet. 1997;349(9066):1641-1649.
12. Hao Q, Tampi M, O'Donnell M, et al. Clopidogrel plus aspirin versus aspirin alone for acute minor ischaemic stroke or high risk transient ischaemic attack: Systematic review and meta-analysis. BMJ. 363:k5108, 2018. doi: 10.1136/bmj.k5108
13. Whelton PK, O'Connell S, Mills KT, He J. Optimal Antihypertensive Systolic Blood Pressure: A Systematic Review and Meta-Analysis. Hypertension. 2024;81(11):2329-2339. doi:10.1161/HYPERTENSIONAHA.124.23597
14. Alexander MJ, Zauner A, Chaloupka JC, et al. WEAVE Trial: Final results in 152 on-label patients. Stroke. 50 (4):889–894, 2019. doi: 10.1161/STROKEAHA.118.023996
15. Kavinsky CJ, Szerlip M, Goldsweig AM, et al. SCAI guidelines for the management of patent foramen ovale. Standards and guidelines. 1 (4), 100039, 2022. doi: 10.1016/j.jscai.2022.100039
16. Lee M, Cheng CY, Wu YL, et al. Association Between Intensity of Low-Density Lipoprotein Cholesterol Reduction With Statin-Based Therapies and Secondary Stroke Prevention: A Meta-analysis of Randomized Clinical Trials. JAMA Neurol. 2022;79(4):349-358. doi:10.1001/jamaneurol.2021.5578
Prognosis for Ischemic Stroke
Stroke severity and progression are often assessed using standardized measures such as the National Institutes of Health (NIH) Stroke Scale (see table ); the score on this scale correlates with extent of functional impairment and prognosis. During the first days, progression and outcome can be difficult to predict. Older age, impaired consciousness, aphasia, and brain stem signs suggest a poor prognosis. Early improvement and younger age suggest a favorable prognosis.

Approximately 50% of patients with moderate or severe hemiplegia and most with milder deficits have a clear sensorium and eventually can take care of their basic needs and walk adequately. Complete neurologic recovery occurs in approximately 10%. Use of the affected limb is usually limited, and most deficits that remain after 12 months are permanent. Patients who have had a stroke are at high risk of subsequent strokes and each tends to worsen neurologic function. Approximately 25% of patients who recover from a first stroke have another stroke within 5 years.
After an ischemic stroke, approximately 20% of patients die in the hospital; mortality rate increases with age.
Key Points
Differentiate ischemic stroke from mimics (eg, postictal paralysis, hemiplegic migraine, CNS infections, functional neurologic disorders).
Although clinical differentiation is imprecise, some clues to help differentiate between common types of stroke include symptom progression (maximal deficits within minutes of onset with embolic versus sometimes stepwise or slow onset with thrombotic), time of onset (day with embolic versus night with thrombotic), and type of deficits (eg, specific syndromes and absence of cortical signs with lacunar infarcts).
Test patients for cardiac causes (including atrial fibrillation) and arterial stenosis (with vascular imaging), and do blood tests (eg, for thrombotic, rheumatic, and other disorders) as indicated.
In general, do not aggressively reduce BP soon after acute ischemic stroke.
To determine eligibility for tPA, use a checklist and, when available, consult an expert, either in person or via telemedicine.
To optimize the salvage of penumbral tissue, begin indicated thrombolytic therapy or mechanical thrombectomy as soon as possible ("time is brain").
To prevent future ischemic strokes, control modifiable risk factors and treat, when appropriate, with antiplatelet therapy, statin therapy, and/or endarterectomy or stenting.
Drug Information for the Topic





