

Acute kidney injury is a rapid decrease in kidney function over days to weeks, causing an accumulation of nitrogenous products in the blood (azotemia) with or without reduction in amount of urine output. It often results from inadequate kidney perfusion due to severe trauma, illness, or surgery but it is sometimes caused by a rapidly progressive, intrinsic kidney disease. Symptoms may include anorexia, nausea, and vomiting. Seizures and coma may occur if the condition is untreated. Fluid, electrolyte, and acid-base disorders develop quickly. Diagnosis is based on laboratory tests of kidney function, including serum creatinine. Urinary indices, urinary sediment examination, and often imaging and other tests (including sometimes a kidney biopsy) are needed to determine the cause. Treatment is directed at the cause but also includes fluid and electrolyte management and sometimes dialysis.
In most cases of acute kidney injury (AKI), creatinine and urea accumulate in the blood over several days, often leading to fluid and electrolyte abnormalities (1). The most serious complications of this process are hyperkalemia and pulmonary edema. Phosphate retention leads to hyperphosphatemia. Hypocalcemia is thought to occur because the impaired kidney no longer produces calcitriol (reducing calcium absorption from the gastrointestinal tract) and because hyperphosphatemia causes calcium phosphate precipitation in the tissues. (reducing calcium absorption from the gastrointestinal tract) and because hyperphosphatemia causes calcium phosphate precipitation in the tissues.Acidosis develops because hydrogen ions cannot be adequately excreted, primarily due to impaired ammonium and bicarbonate production in the proximal tubules (2). With significant uremia, coagulation may be impaired, and encephalopathy and pericarditis may develop. Urine output varies with the type and cause of AKI.
General references
1. Kellum JA, Lameire N; KDIGO AKI Guideline Work Group. Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (Part 1). Crit Care. 2013;17(1):204. Published 2013 Feb 4. doi:10.1186/cc11454
2. Nagami GT, Kraut JA. Regulation of Acid-Base Balance in Patients With Chronic Kidney Disease. Adv Chronic Kidney Dis. 2022;29(4):337-342. doi:10.1053/j.ackd.2022.05.004
Etiology of AKI
Causes of acute kidney injury (AKI; see table ) can be classified as follows (1, 2):
Prerenal
Renal (intrinsic)
Postrenal
Prerenal AKI is due to inadequate renal perfusion. The main causes are
Extracellular fluid volume depletion (eg, due to inadequate fluid intake, diarrheal illness, sepsis, severe trauma, surgery, hemorrhage)
Cardiovascular disease (eg, heart failure, cardiogenic shock)
Decompensated liver disease
Medications that reduce renal perfusion (eg, nonsteroidal anti-inflammatory drugs, angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers, calcineurin inhibitors)Medications that reduce renal perfusion (eg, nonsteroidal anti-inflammatory drugs, angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers, calcineurin inhibitors)
Prerenal conditions typically do not cause permanent kidney damage (and hence are potentially reversible) unless hypoperfusion is severe and/or prolonged. Hypoperfusion of an otherwise functioning kidney leads to enhanced reabsorption of sodium and water, resulting in oliguria (urine output < 500 mL/day) with high urine osmolality and low urine sodium.
Renal causes of AKI involve intrinsic kidney disease or damage. Disorders may involve the blood vessels, glomeruli, tubules, or interstitium. The most common causes are
Nephrotoxins (including prescription and over-the-counter medications—see Analgesic Nephropathy) (3, 4)

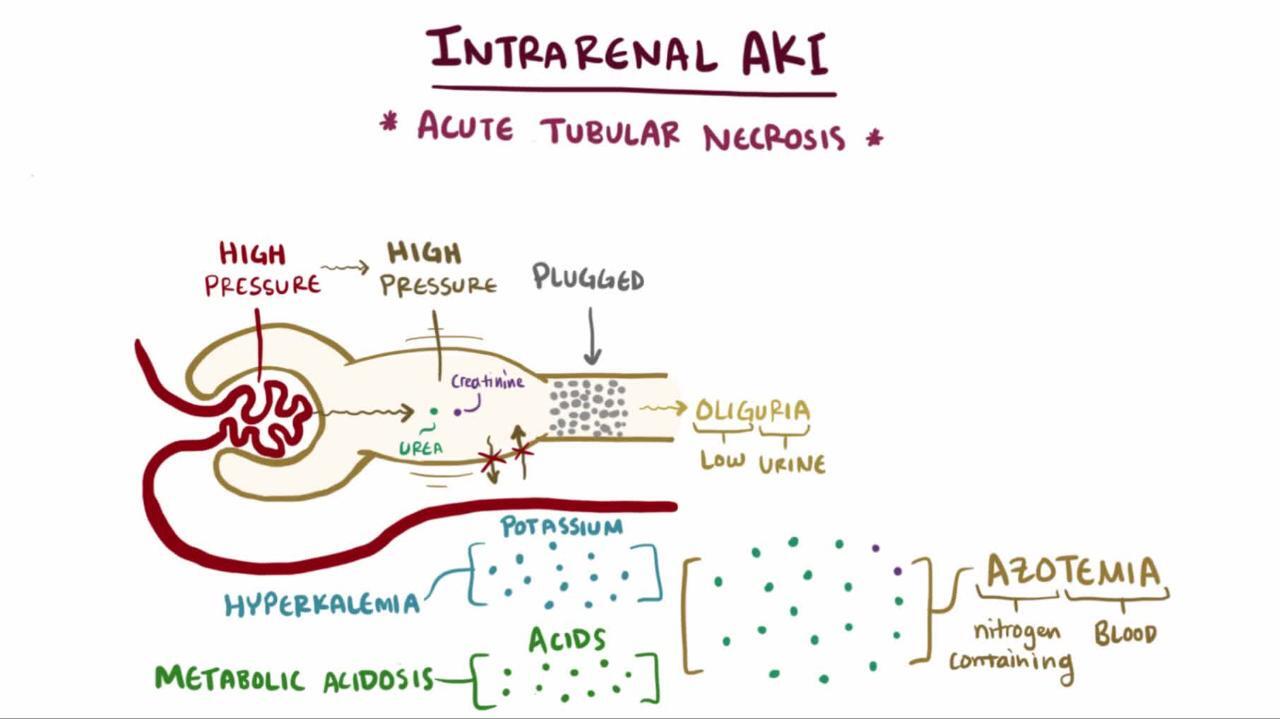
Acute tubular necrosis (ATN) is a form of kidney injury affecting the renal tubules, most commonly caused by ischemia. The other major cause of ATN is nephrotoxic injury from agents, which may result from medications (eg, vancomycin, aminoglycosides), iodinated radiocontrast media, and endogenous pigments such as myoglobin from rhabdomyolysis and hemoglobin from hemolysis. These mechanisms of tubular damage are complex and interdependent, rendering the term (ATN) is a form of kidney injury affecting the renal tubules, most commonly caused by ischemia. The other major cause of ATN is nephrotoxic injury from agents, which may result from medications (eg, vancomycin, aminoglycosides), iodinated radiocontrast media, and endogenous pigments such as myoglobin from rhabdomyolysis and hemoglobin from hemolysis. These mechanisms of tubular damage are complex and interdependent, rendering the termacute tubular necrosis an imprecise description.
Interstitial inflammation (acute tubulointerstitial nephritis) usually involves an immunologic or allergic phenomenon.
Glomerular disease is characterized by damage to the glomerular filtration barrier with resultant reduction in glomerular filtration rate (GFR) and increased glomerular capillary permeability to proteins and/or red blood cells. Glomerular diseases include inflammatory conditions such as glomerulonephritis and noninflammatory conditions such as minimal change disease and focal segmental glomerulosclerosis.
Various infections can result in intrinsic AKI. Postinfectious glomerulonephritis is caused by nephritogenic strains of group A beta-hemolytic streptococci. Tubular injury has also been identified histopathologically after COVID-19–associated kidney injury (5). Any viral or bacterial infections leading to sepsis can cause AKI due to systemic inflammation and/or shock.
Postrenal AKIis caused by obstruction of the urinary tract, which can occur at any level of the urinary tract from the renal tubules to the urethra (obstructive uropathy). Obstruction can also occur on the microscopic level within the tubules when crystalline or proteinaceous material precipitates.
Obstructed ultrafiltrate, in tubules or more distally, increases pressure in the urinary space of the glomerulus, reducing GFR. AKI recovery after relief of obstruction is more likely with shorter duration of obstruction, and may be irreversible in prolonged (weeks to months) cases (6).
To produce significant AKI, obstruction at the level of the ureter requires involvement of both ureters unless the patient has only a single functioning kidney.
Bladder outlet obstruction due to an enlarged prostate is probably the most common cause of sudden, and sometimes total, cessation of urinary output in men.
Major Causes of Acute Kidney Injury
Cause | Examples |
|---|---|
Prerenal | |
ECF volume depletion | Excessive diuresis Gastrointestinal losses Hemorrhage Loss of intravascular fluid into the extravascular space (due to ascites, peritonitis, pancreatitis, or burns) Loss of fluid from skin and mucous membranes Renal salt- and water-wasting states |
Low cardiac output | Cardiomyopathy with reduced left ventricular systolic function Myocardial infarction with cardiogenic shock or exacerbation of underlying heart failure |
Low systemic vascular resistance | Antihypertensive medications Liver failure |
Vasoconstriction of afferent and glomerular arterioles | Calcineurin inhibitors (eg, cyclosporine, tacrolimus)Calcineurin inhibitors (eg, cyclosporine, tacrolimus) NSAIDs |
Decreased efferent arteriolar tone (leading to decreased GFR from reduced glomerular transcapillary pressure, especially in patients with bilateral renal artery stenosis) | ACE inhibitors ARBs |
Renal | |
Acute tubular injury | Ischemia (prolonged or severe prerenal state):
Toxins:
|
Acute glomerulonephritis | ANCA-associated:
Immune-complex:
|
Acute tubulointerstitial nephritis | Drug reaction (eg, beta-lactams, proton-pump inhibitors, immune checkpoint inhibitors, NSAIDs, sulfonamides, ciprofloxacin, thiazide diuretics, furosemide, cimetidine, phenytoin, allopurinol)Drug reaction (eg, beta-lactams, proton-pump inhibitors, immune checkpoint inhibitors, NSAIDs, sulfonamides, ciprofloxacin, thiazide diuretics, furosemide, cimetidine, phenytoin, allopurinol) Papillary necrosis |
Acute vascular nephropathy | Atheroembolism Thrombotic microangiopathies |
Infiltrative diseases | Leukemia Lymphoma |
Postrenal | |
Tubular precipitation | AcyclovirAcyclovir Calcium oxalate (due to ingestion of ethylene glycol or excessive vitamin C) IndinavirIndinavir MethotrexateMethotrexate Myeloma protein Myoglobin Sulfonamides TriamtereneTriamterene Uric acid (tumor lysis) |
Ureteral obstruction | Intrinsic:
Extrinsic:
|
Bladder obstruction | Mechanical:
Neurogenic:
|
ACE = angiotensin-converting enzyme; ANCA = antineutrophil cytoplasmic antibody; ARB = angiotensin II receptor blocker; ECF angiotensin II receptor blocker; ECF= extracellular fluid; GBM = glomerular basement membrane; GFR = glomerular filtration rate; NSAIDs = nonsteroidal anti-inflammatory drugs. | |
Etiology references
1. Lameire NH, Bagga A, Cruz D, et al. Acute kidney injury: an increasing global concern. Lancet. 2013;382(9887):170-179. doi:10.1016/S0140-6736(13)60647-9
2. Turgut F, Awad AS, Abdel-Rahman EM. Acute Kidney Injury: Medical Causes and Pathogenesis. J Clin Med. 2023;12(1):375. Published 2023 Jan 3. doi:10.3390/jcm12010375
3. Hosohata K, Inada A, Oyama S, Furushima D, Yamada H, Iwanaga K. Surveillance of drugs that most frequently induce acute kidney injury: A pharmacovigilance approach. J Clin Pharm Ther. 2019;44(1):49-53. doi:10.1111/jcpt.12748
4. Pierson-Marchandise M, Gras V, Moragny J, et al. The drugs that mostly frequently induce acute kidney injury: a case - noncase study of a pharmacovigilance database. Br J Clin Pharmacol. 2017;83(6):1341-1349. doi:10.1111/bcp.13216
5. Legrand M, Bell S, Forni L, et al. Pathophysiology of COVID-19-associated acute kidney injury. Nat Rev Nephrol. 2021;17(11):751-764. doi:10.1038/s41581-021-00452-0
6. Gembillo G, Spadaro G, Santoro D. Link between obstructive uropathy and acute kidney injury. World J Nephrol. 2025 Mar 25;14(1):99120. doi: 10.5527/wjn.v14.i1.99120.
Symptoms and Signs of AKI
Initially, decreased urine output may be the only sign of AKI, followed by weight gain and peripheral edema. Often, the predominant symptoms are those of the underlying illness (eg, fever and hypotension in a patient with sepsis) that precipitated renal deterioration.
Symptoms of uremia may develop later as nitrogenous products accumulate. Such symptoms include
Anorexia
Nausea
Vomiting
Weakness
Myoclonic jerks
Seizures
Confusion
Coma
Asterixis and hyperreflexia may be present on examination. Chest pain (typically worse with inspiration or when recumbent), a pericardial friction rub, and findings of pericardial tamponade may occur if uremic pericarditis is present. Fluid accumulation in the lungs may cause dyspnea and crackles on auscultation.

Other findings depend on the cause. Urine may be dark brown, reddish brown, or tea colored in glomerulonephritis or myoglobinuria. A palpable bladder may be present with outlet obstruction. The costovertebral angle may be tender if the kidney is acutely enlarged due to hydronephrosis from ureteral obstruction.
Changes in urine output
Amount of urine output during acute kidney injury (AKI) does not clearly differentiate between prerenal, renal, or postrenal causes. However, measurement of urine output can allow for earlier detection of AKI before serum creatinine rise, and risk stratification, and prognosis of AKI (with low urine output posing a high risk) (1, 2).
In acute kidney injury, urine output may have 3 phases:
The prodromal phase usually has normal urine output and varies in duration depending on causative factors (eg, the amount of toxin ingested, the duration and severity of hypotension).
The oliguric phase has urine output typically between 50 and 500 mL/day. The duration of the oliguric phase is unpredictable, depending on etiology of AKI and time to treatment. However, many patients are never oliguric. Nonoliguric patients have lower mortality and morbidity and less need for dialysis.
In the postoliguric phase, urine output gradually returns to normal, but serum creatinine and urea levels may not fall for several more days. Tubular dysfunction may persist for a few days or weeks and is manifested by sodium wasting, polyuria (possibly massive) unresponsive to vasopressin, or hyperchloremic metabolic acidosis.
Symptoms and signs references
1. Kellum JA, Sileanu FE, Murugan R, Lucko N, Shaw AD, Clermont G. Classifying AKI by Urine Output versus Serum Creatinine Level. J Am Soc Nephrol. 2015 Sep;26(9):2231-8. doi: 10.1681/ASN.2014070724
2. Parker RA, Himmelfarb J, Tolkoff-Rubin N, Chandran P, Wingard RL, Hakim RM. Prognosis of patients with acute renal failure requiring dialysis: results of a multicenter study. Am J Kidney Dis. 1998 Sep;32(3):432-43. doi: 10.1053/ajkd.1998.v32.pm9740160
Diagnosis of AKI
History and physical examination, including review of prescription and over-the-counter medications and exposure to iodinated IV contrast
Laboratory tests, including serum creatinine, blood urea nitrogen (BUN), electrolytes Laboratory tests, including serum creatinine, blood urea nitrogen (BUN), electrolytes
Urinary studies, including urinalysis; urinary sediment; urine sodium, urea, protein, and creatinine concentrationUrinary studies, including urinalysis; urinary sediment; urine sodium, urea, protein, and creatinine concentration
Urinary diagnostic indices (eg, fractional excretion of sodium)
Postvoid residual bladder volume and/or renal ultrasound if postrenal cause suspected
Acute kidney injury (AKI) is suspected when urine output falls or serum blood urea nitrogen (BUN) and creatinine rise.
Per the KDIGO (Kidney Disease: Improving Global Outcomes) Clinical Practice Guideline for Acute Kidney Injury (1), AKI is defined as any of the following:
Increase in the serum creatinine value of ≥ 0.3 mg/dL (26.5 micromol/L) in 48 hours
Increase in serum creatinine of ≥ 1.5 times baseline within the prior 7 days
Urine volume < 0.5 mL/kg/hour for 6 hours
Evaluation should determine the presence and type of AKI and seek a cause. Blood tests generally include complete blood count (CBC), blood urea nitrogen (BUN), creatinine, and electrolytes (including calcium and phosphate) (2). Urine tests include sodium, urea, protein, and creatinine concentration; and microscopic analysis of sediment. Early detection and treatment increase the chances of reversing renal injury and, in some cases, preventing progression to the need for dialysis.
A progressive daily rise in serum creatinine is diagnostic of AKI. Serum creatinine can increase by as much as 2 mg/dL/day (180 micromol/L/day), depending on the amount of creatinine produced (which varies with lean body mass) and total body water.
Blood urea nitrogen may increase by 10 to 20 mg/dL/day (3.6 to 7.1 mmol urea/L/day), but BUN may be misleading because it is frequently elevated in response to increased protein catabolism resulting from surgery, trauma, glucocorticoids, burns, transfusion reactions, parenteral nutrition, or gastrointestinal or other internal bleeding.
When creatinine is rising, 24-hour urine collection for creatinine clearance and the various formulas used to calculate creatinine clearance from serum creatinine are inaccurate and should not be used in estimating the glomerular filtration rate (eGFR), because the rise in serum creatinine concentration is a delayed function of GFR decline.
Other laboratory findings are
Progressive acidosis: Acidosis is ordinarily moderate, with a plasma bicarbonate content of 15 to 20 mmol/L; however, acidosis may be severe if there is underlying sepsis or tissue ischemia.
Hyperkalemia : Rise in serum potassium concentration depends on overall metabolism, dietary intake, medications, and potential tissue necrosis or cellular lysis.
Hyponatremia: Hyponatremia usually is moderate (serum sodium, 125 to 135 mmol/L) and correlates with a surplus of dietary or intravenous water intake.
Anemia: Normochromic-normocytic anemia with a hematocrit of 25 to 30% is typical.
Hyperphosphatemia and hypocalcemia are common in AKI and may be profound in patients with rhabdomyolysis or tumor lysis syndrome. Profound hypocalcemia in rhabdomyolysis apparently results from the combined effects of calcium deposition in necrotic muscle, reduced calcitriol production, resistance of bone to parathyroid hormone (PTH), and hyperphosphatemia. During recovery from AKI following rhabdomyolysis-induced production, resistance of bone to parathyroid hormone (PTH), and hyperphosphatemia. During recovery from AKI following rhabdomyolysis-inducedacute tubular necrosis, hypercalcemia may supervene as renal calcitriol production increases, the bone becomes responsive to PTH, and calcium deposits are mobilized from damaged tissue. Hypercalcemia during recovery from AKI is otherwise uncommon.
Determination of cause
Immediately reversible prerenal or postrenal causes of acute kidney injury must be excluded first. Extracellular fluid (ECF) volume depletion and obstruction are considered in all patients. The medication history must be accurately reviewed and all potentially renal toxic agents stopped. Urinary diagnostic indices (see table ) may be helpful in distinguishing prerenal AKI from acute tubular injury in an oliguric state, which are the most common causes of AKI in hospitalized patients. Urinary diagnostic indices are not reliable in nonoliguric states (3).
Prerenal causes are often apparent clinically. If so, correction of an underlying hemodynamic abnormality should be attempted. For example, in hypovolemia, volume infusion can be tried; in heart failure (HF), diuretics and afterload-reducing medications can be tried. Abatement of AKI confirms a prerenal cause.
Urinary Diagnostic Indices in Prerenal Acute Kidney Injury and Acute Tubular Injury
Index | Prerenal | Tubular Injury |
|---|---|---|
U/P osmolality | > 1.5 | 1–1.5 |
Urine sodium (mmol/L) | < 10 | > 40 |
Fractional excretion of sodium (FENa)* | < 1% | > 1% |
BUN/creatinine ratio | > 20 | < 10 |
* U/P Na ÷ U/P creatinine. | ||
BUN = blood urea nitrogen; U/P = urine-to-plasma ratio. | ||
Adapted from Miller TR, Anderson RJ, Linas SL, et al. Urinary diagnostic indices in acute renal failure. Ann Intern Med. 89(1):47-50, 1978; used with permission of the American College of Physicians and the author. | ||

Postrenal causes should be sought in most cases of AKI. Immediately after the patient voids, bedside ultrasound of the bladder is performed (or, alternatively, a urinary catheter is placed) to determine the residual urine in the bladder. A postvoid residual urine volume > 100 mL suggests inadequate bladder emptying (eg, due to bladder outlet obstruction or detrusor muscle weakness and neurogenic bladder). The catheter, if placed, may be kept in to accurately monitor urine output in response to therapies, but the catheter is removed in patients who are anuric (if bladder outlet obstruction is not present) to decrease the risk of infection.
Renal ultrasound can also be obtained to identify a more proximal obstruction. However, ultrasound may not detect an obstructive pathology because the collecting system is not always dilated, especially when the condition is acute, the ureter is encased (eg, in retroperitoneal fibrosis or neoplasm), or the patient has concomitant hypovolemia. If obstruction is strongly suspected, noncontrast CT can establish the site of obstruction and guide therapy.
The urinary sediment may provide etiologic clues. A normal urine sediment occurs in prerenal AKI and sometimes in obstructive uropathy. With renal tubular injury, the sediment characteristically contains tubular cells, tubular cell casts, and many granular casts (often with brown pigmentation). Urinary eosinophils may indicate allergic tubulointerstitial nephritis, but the diagnostic accuracy of this finding is limited. Red blood cell (RBC) casts and dysmorphic RBCs indicate glomerulonephritis or vasculitis but rarely may occur in acute tubular necrosis.
Renal causes are sometimes suggested by clinical findings. Patients with glomerulonephritis often have edema, marked proteinuria (nephrotic syndrome), or signs of arteritis in the skin and retina, often without a history of intrinsic renal disease. Hemoptysis may result from granulomatosis with polyangiitis or anti-GBM disease (Goodpasture syndrome). Certain rashes (eg, erythema nodosum, cutaneous vasculitis, discoid lupus) may indicate cryoglobulinemia, systemic lupus erythematosus (SLE), or immunoglobulin A-associated vasculitis. Tubulointerstitial nephritis, drug allergy, and possibly microscopic polyangiitis are suggested by a history of drug ingestion and a maculopapular or purpuric rash.
To further differentiate renal causes, antistreptolysin-O and complement titers, antinuclear antibodies, and antineutrophil cytoplasmic antibodies are determined.
Renal biopsy may be done if the diagnosis remains elusive (see table ).
Causes of Acute Kidney Injury Based on Laboratory Findings
Blood Test | Finding | Possible Diagnosis |
|---|---|---|
Antiglomerular basement membrane (anti-GBM) antibodies | Positive | Anti-GBM disease (Goodpasture syndrome) |
Antineutrophil cytoplasmic antibodies | Positive | Small-vessel vasculitis (eg, granulomatosis with polyangiitis or microscopic polyangiitis) |
Antinuclear antibodies or antibodies to double-stranded DNA | Positive | |
Antistreptolysin-O or antibodies to streptokinase or hyaluronidaseAntistreptolysin-O or antibodies to streptokinase or hyaluronidase | Positive | |
Creatine kinase or myoglobin level | Markedly elevated | |
Complement titers | Low | Cholesterol embolization Postinfectious glomerulonephritis SLE |
Protein electrophoresis (serum) | Monoclonal spike | |
Uric acid level | Elevated | Cancer or tumor lysis syndrome (leading to uric acid crystals) Prerenal acute kidney injury |
Imaging
In addition to renal ultrasound, other imaging tests are occasionally obtained. In evaluating for ureteral obstruction, noncontrast CT is preferred over antegrade and retrograde urography. In addition to its ability to delineate soft-tissue structures and calcium-containing calculi, CT can detect nonradiopaque calculi.
Iodinated contrast agents should be avoided if possible. However, conventional renal arteriography may occasionally be indicated if macrovascular causes are suggested clinically, and intervention is anticipated (eg, renal artery stenosis). Magnetic resonance angiography can be used for diagnosing renal artery stenosis as well as thrombosis of both arteries and veins because gadolinium is used as the contrast agent, which has a lower risk of AKI than the iodinated contrast agents used in angiography and contrast-enhanced CT. However, gadolinium is involved in the pathogenesis of nephrogenic systemic fibrosis, a serious complication that occurs in patients with AKI as well as chronic kidney disease. Thus, gadolinium should be avoided if possible in patients with renal function below an estimated glomerular filtration rate (eGFR) of 30 mL/minute/1.73m2. If clinically indicated, then group II gadolinium agents should be used preferentially due to lower risk of nephrogenic systemic fibrosis. (See Disadvantages of MRI.)
Kidney size, as determined with imaging tests, is helpful to know because, a normal or enlarged kidney favors reversibility, whereas a small kidney suggests chronic renal insufficiency. However, some chronic kidney diseases tend to present with enlarged kidneys, including the following:
Staging of AKI
Once the patient's volume status is optimized and genitourinary obstruction is excluded, AKI can be classified into 3 stages based on serum creatinine level or the amount of urine output (see table ). The staging criteria provide a useful framework for risk stratification and for guiding management, but clinical decisions should not be dictated by stage alone.
Staging Criteria for Acute Kidney Injury (KDIGO 2012)*
Stage | AKI measures (any of the below for each stage) | ||
|---|---|---|---|
Rise in serum creatinine | Decline in the amount of urine output | Renal replacement therapy | |
1 | ≥ 0.3 mg/dL (26.52 micromol/L) or 1.5–1.9 times baseline | < 0.5 mL/kg/hour for 6–12 hours | Not indicated |
2 | 2–2.9 times baseline | < 0.5 mL/kg/hour for ≥ 12 hours | Not indicated |
3 | ≥ 4.0 mg/dL (353.60 micromol/L) or ≥ 3 times baseline | < 0.3 mL/kg/hour for ≥ 24 hours or anuria for ≥ 12 hours | Indicated |
* Data from KDIGO (Kidney Disease: Improving Global Outcomes) Acute Kidney Injury Work Group: KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int. Suppl. 2:1-138, 2012. | |||
Diagnosis references
1. KDIGO (Kidney Disease: Improving Global Outcomes) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int. Suppl. 2:1-138, 2012.
2. Ostermann M, Lumlertgul N, Jeong R, See E, Joannidis M, James M. Acute kidney injury. Lancet. 2025;405(10474):241-256. doi:10.1016/S0140-6736(24)02385-7
3. Hamadah A, Gharaibeh K. Fractional Excretion of Sodium and Urea are Useful Tools in the Evaluation of AKI: PRO. . Fractional Excretion of Sodium and Urea are Useful Tools in the Evaluation of AKI: PRO.Kidney360. 2023 Jun 1;4(6):e725-e727. doi: 10.34067/KID.0002492022
Treatment of AKI
Immediate treatment of pulmonary edema and hyperkalemia
Dialysis as needed to control hyperkalemia, pulmonary edema, metabolic acidosis, and uremic symptoms
Adjustment of medication regimen for degree of renal dysfunction
Usually restriction of water, sodium, phosphate, and potassium intake, but provision of adequate protein
Possibly phosphate binders (for hyperphosphatemia) and intestinal potassium binders (for hyperkalemia)
Emergency treatment
Life-threatening complications are addressed, preferably in a critical care unit. Pulmonary edema is treated with oxygen, IV vasodilators (eg, nitroglycerin), diuretics (often ineffective in AKI), or is treated with oxygen, IV vasodilators (eg, nitroglycerin), diuretics (often ineffective in AKI), ordialysis.
Hyperkalemia is treated as needed with IV infusion of 10 mL of 10% calcium gluconate, 50 g of dextrose, and 5 to 10 units of insulin. These medications do not reduce total body potassium, so further (but slower-acting) treatment is needed (eg, intestinal potassium binders, diuretics, dialysis). is treated as needed with IV infusion of 10 mL of 10% calcium gluconate, 50 g of dextrose, and 5 to 10 units of insulin. These medications do not reduce total body potassium, so further (but slower-acting) treatment is needed (eg, intestinal potassium binders, diuretics, dialysis).
Although correction of mild to moderate metabolic acidosis with sodium bicarbonate is controversial, IV sodium bicarbonate for the correction of severe Although correction of mild to moderate metabolic acidosis with sodium bicarbonate is controversial, IV sodium bicarbonate for the correction of severemetabolic acidosis (pH < 7.20) is often recommended to decrease mortality and need for dialysis treatment (1, 2). For patients without volume overload, bicarbonate can be administered as an infusion of isotonic bicarbonate (eg, 150 mEq of sodium bicarbonate in 1 liter of 5% dextrose) at a rate of 50 to 200 mL/hour. For patients with volume overload or a life-threatening condition (eg, cardiac arrest), a highly concentrated hypertonic bicarbonate (50 mEq of sodium bicarbonate in 50 mL) is administered over 1 to 2 minutes. Because variations in body buffer systems and the rate of acid production are highly variable, calculating the amount of bicarbonate needed to achieve a full correction is usually not recommended. Instead, the amount of sodium bicarbonate is titrated based on reassessment of serial laboratory tests when monitoring acidosis.). For patients without volume overload, bicarbonate can be administered as an infusion of isotonic bicarbonate (eg, 150 mEq of sodium bicarbonate in 1 liter of 5% dextrose) at a rate of 50 to 200 mL/hour. For patients with volume overload or a life-threatening condition (eg, cardiac arrest), a highly concentrated hypertonic bicarbonate (50 mEq of sodium bicarbonate in 50 mL) is administered over 1 to 2 minutes. Because variations in body buffer systems and the rate of acid production are highly variable, calculating the amount of bicarbonate needed to achieve a full correction is usually not recommended. Instead, the amount of sodium bicarbonate is titrated based on reassessment of serial laboratory tests when monitoring acidosis.
Hemodialysis or hemofiltration is initiated when
Severe electrolyte abnormalities cannot otherwise be controlled (eg, potassium > 6 mmol/L)
Pulmonary edema persists despite treatment with medication
Metabolic acidosis is unresponsive to treatment
Uremic symptoms occur (eg, vomiting thought to be due to uremia, asterixis, encephalopathy, pericarditis, seizures)
Blood urea nitrogen (BUN) and creatinine levels alone are not reliable indicators for determining when to initiate dialysis in acute kidney injury (AKI). In asymptomatic patients who are not seriously ill and likely to recover, dialysis can be deferred until (1) uremic symptoms (eg, encephalopathy, pericarditis) occur, or (2) electrolyte and/or volume complications are not amenable to medical therapies. This approach avoids the placement of a central venous catheter with its attendant complications and costs.
General measures
Nephrotoxic medications are discontinued, and all medications excreted by the kidneys (eg, digoxin, some antibiotics) are adjusted; serum levels are useful.Nephrotoxic medications are discontinued, and all medications excreted by the kidneys (eg, digoxin, some antibiotics) are adjusted; serum levels are useful.
Daily water intake is restricted to a volume equal to the previous day’s urine output plus measured extrarenal losses (eg, nasogastric tube suction fluid, vomitus, diarrhea) plus 500 to 1000 mL/day for insensible loss. Water intake can be further restricted for hyponatremia or increased for hypernatremia. Although weight gain indicates excess fluid, water intake is not decreased if serum sodium remains normal; instead, dietary sodium is restricted.
Sodium intake and potassium intake are minimized except in patients with prior deficiencies or gastrointestinal losses. An adequate diet should be provided, including daily protein intake of about 0.8 g/kg. If oral or enteral nutrition is impossible, parenteral nutrition is used; however, in AKI, risks of fluid overload, hyperosmolality, and infection are increased by IV nutrition. Calcium salts (calcium carbonate, calcium acetate) or iron-based or synthetic phosphate binders before meals help maintain serum phosphate at Sodium intake and potassium intake are minimized except in patients with prior deficiencies or gastrointestinal losses. An adequate diet should be provided, including daily protein intake of about 0.8 g/kg. If oral or enteral nutrition is impossible, parenteral nutrition is used; however, in AKI, risks of fluid overload, hyperosmolality, and infection are increased by IV nutrition. Calcium salts (calcium carbonate, calcium acetate) or iron-based or synthetic phosphate binders before meals help maintain serum phosphate at< 5.5 mg/dL (< 1.8 mmol/L).
If needed to help maintain serum potassium at < 6 mmol/L in the absence of dialysis (eg, if other therapies, such as diuretics, fail to lower potassium), a cation-exchange resin is prescribed when delayed onset of action of several hours is acceptable. Sodium polystyrene sulfonate is available in oral or rectal formulation, but it should be avoided in patients at high risk for bowel dysmotility (bowel obstruction, impaction, post-operative ileus); patiromer and sodium zirconium cyclosilicate are available via the oral route only.(eg, if other therapies, such as diuretics, fail to lower potassium), a cation-exchange resin is prescribed when delayed onset of action of several hours is acceptable. Sodium polystyrene sulfonate is available in oral or rectal formulation, but it should be avoided in patients at high risk for bowel dysmotility (bowel obstruction, impaction, post-operative ileus); patiromer and sodium zirconium cyclosilicate are available via the oral route only.
An indwelling bladder catheter is rarely needed and should be used only when necessary because of an increased risk of urinary tract infection and urosepsis.
In many patients, a brisk and even dramatic diuresis after relief of obstruction is a physiologic response to the expansion of extracellular fluid (ECF) during obstruction and does not compromise volume status. However, polyuria accompanied by the excretion of large amounts of sodium, potassium, magnesium, and other solutes may cause hypokalemia, hyponatremia, hypernatremia (if free water is not provided), hypomagnesemia, or marked contraction of ECF volume with peripheral vascular collapse. In this postoliguric phase, close attention to fluid and electrolyte balance is mandatory. Overzealous administration of salt and water after relief of obstruction can prolong diuresis. When postoliguric diuresis occurs, replacement of urine output with 0.45% saline at about 75% of urine output prevents volume depletion and the tendency for excessive free water loss while allowing the body to eliminate excessive volume if this is the cause of the polyuria.
Treatment references
1. Jung B, Jabaudon M, De Jong A, et al. Sodium Bicarbonate for Severe Metabolic Acidemia and Acute Kidney Injury: The BICARICU-2 Randomized Clinical Trial. . Sodium Bicarbonate for Severe Metabolic Acidemia and Acute Kidney Injury: The BICARICU-2 Randomized Clinical Trial.JAMA. 2025;334(22):2000-2010. doi:10.1001/jama.2025.20231
2. Jaber S, Paugam C, Futier E, et al. Sodium bicarbonate therapy for patients with severe metabolic acidaemia in the intensive care unit (BICAR-ICU): a multicentre, open-label, randomised controlled, phase 3 trial. . Sodium bicarbonate therapy for patients with severe metabolic acidaemia in the intensive care unit (BICAR-ICU): a multicentre, open-label, randomised controlled, phase 3 trial.Lancet. 2018;392(10141):31-40. doi:10.1016/S0140-6736(18)31080-8
Prognosis for AKI
Prognosis for recovery of renal function after acute kidney injury (AKI) correlates with premorbid kidney function. Patients with underlying chronic kidney disease (CKD) are at greater risk of developing AKI, requiring dialysis for treatment of AKI, and progressing to end-stage kidney disease (ESKD).
Prognosis of nonoliguric AKI (urine output > 500 mL/day) is better than that of oliguric or anuric AKI (1). Increase in urine output with or without aid of a diuretic suggests renal function recovery or less severe AKI. After hospital admission, even after recovery from AKI (eg, normal post-discharge kidney function), patients are still at risk for future CKD and ESKD (2).
In addition, both the presence and severity of AKI are risk factors for increased mortality in hospitalized patients. Among a group of Medicare beneficiaries hospitalized in the United States in 2023 without AKI, death or discharge to hospice occurred in 5.4% of admissions without AKI, 13.8% of admissions with AKI not requiring dialysis, and 38.3% of admissions requiring dialysis (3). In another cohort of adults ≥ 65 who received outpatient dialysis after hospital discharge, over half were still dependent on dialysis at 6 months.
Prognosis references
1. Parker RA, Himmelfarb J, Tolkoff-Rubin N, Chandran P, Wingard RL, Hakim RM. Prognosis of patients with acute renal failure requiring dialysis: results of a multicenter study. Am J Kidney Dis. 1998 Sep;32(3):432-43. doi: 10.1053/ajkd.1998.v32.pm9740160. PMID: 9740160.
2. Sawhney S, Marks A, Fluck N, Levin A, McLernon D, Prescott G, Black C. Post-discharge kidney function is associated with subsequent ten-year renal progression risk among survivors of acute kidney injury. Kidney Int. 2017 Aug;92(2):440-452. doi: 10.1016/j.kint.2017.02.019
3. National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), United States Renal Data System (USRDS). Acute Kidney Injury. Accessed November 23, 2025.
Prevention of AKI
Acute kidney injury (AKI) can often be prevented by maintaining normal fluid balance, blood volume, and blood pressure in patients with trauma, burns, or severe hemorrhage and in those undergoing major surgery. Infusion of isotonic saline and blood may be helpful.
Use of iodinated contrast agents should be minimized, particularly in at-risk groups (eg, older patients and those with preexisting renal insufficiency [eg, GFR < 30 mL/min/1.73m2], volume depletion, diabetes, or heart failure) for contrast nephropathy. If contrast agents are necessary, risk can be lowered by minimizing volume of the IV contrast agent, using nonionic and low osmolal or iso-osmolal contrast agents, avoiding nonsteroidal anti-inflammatory drugs, and pretreating with normal saline before , during, and after the test. Infusion of isotonic sodium bicarbonate has also been used successfully instead of normal saline, especially in patients with concurrent . If contrast agents are necessary, risk can be lowered by minimizing volume of the IV contrast agent, using nonionic and low osmolal or iso-osmolal contrast agents, avoiding nonsteroidal anti-inflammatory drugs, and pretreating with normal saline before , during, and after the test. Infusion of isotonic sodium bicarbonate has also been used successfully instead of normal saline, especially in patients with concurrentmetabolic acidosis.
Tumor lysis syndrome may occur in patients treated with cytolytic therapy for neoplastic diseases (eg, lymphoma, leukemia), and can result in significant increases in serum uric acid, potassium, and phosphorus (1). To prevent AKI in patients at high risk for tumor lysis syndrome, treatment with rasburicase or allopurinol to lower uric acid levels should be considered along with increasing oral or IV fluids to increase urine output and reduce urate crystalluria. ). To prevent AKI in patients at high risk for tumor lysis syndrome, treatment with rasburicase or allopurinol to lower uric acid levels should be considered along with increasing oral or IV fluids to increase urine output and reduce urate crystalluria.
Prevention reference
1. Rosner MH, Perazella MA. Acute Kidney Injury in Patients with Cancer. N Engl J Med. 2017;376(18):1770-1781. doi:10.1056/NEJMra1613984
Key Points
Causes of AKI can be prerenal (eg, kidney hypoperfusion), renal (eg, direct effects on the kidney), or postrenal (eg, urinary tract obstruction distal to the kidneys).
With AKI, consider ECF volume depletion and nephrotoxins, obtain urinary diagnostic indices and measure bladder residual volume to identify obstruction.
Avoid or minimize use of iodinated IV contrast in imaging studies.
Initiate hemodialysis or hemofiltration as needed for pulmonary edema, hyperkalemia, metabolic acidosis, or uremic symptoms unresponsive to other treatments.
Minimize risk of AKI in patients at risk by maintaining normal fluid balance, avoiding nephrotoxins (including iodinated intravenous contrast agents) when possible, and taking precautions such as giving fluids or medications when contrast or cytolytic therapy is necessary.
Drug Information for the Topic





