

Postinfectious glomerulonephritis occurs after infection, usually with a nephritogenic strain of group A beta-hemolytic streptococcus. Diagnosis is suggested by history and urinalysis and confirmed by finding a low complement level and sometimes by antibody testing. Treatment is supportive. Prognosis is excellent.
Etiology of Postinfectious Glomerulonephritis
Postinfectious glomerulonephritis (PIGN), a nephritic syndrome, is the most common cause of acute glomerulonephritis in children 3 to 15 years old. It is more common in highly populated and economically disadvantaged areas (1).
Most cases are caused by nephritogenic strains of group A beta-hemolytic streptococci, most notably type 12 (which causes pharyngitis) and type 49 (which causes impetigo). A latency period of 6 to 21 days between infection and glomerulonephritis onset is typical, but latency may extend up to 6 weeks. This is longer than the latency period between immunoglobulin A nephropathy and its antecedent infection.
PIGN associated with staphylococcal infections is growing in incidence and affects an older population (1).
Less common pathogens are other nonstreptococcal bacteria, viruses, parasites, rickettsiae, and fungi (see table ). Bacterial endocarditis and ventriculoatrial shunt infections are additional important conditions in which PIGN develops; ventriculoperitoneal shunts are more resistant to infection.
The mechanism is unknown, but microbial antigens are thought to bind to the glomerular basement membrane and activate primarily the alternate complement pathway both directly and via interaction with circulating antibodies, causing glomerular damage, which may be focal or diffuse. Alternatively, circulating immune complexes could precipitate on the glomerular basement membrane.
Etiology reference
1. Satoskar AA, Parikh SV, Nadasdy T. Epidemiology, pathogenesis, treatment and outcomes of infection-associated glomerulonephritis. Nat Rev Nephrol 2020;16(1):32-50. doi:10.1038/s41581-019-0178-8
Symptoms and Signs of Postinfectious Glomerulonephritis
Symptoms and signs range from asymptomatic hematuria and mild proteinuria to full-blown nephritis with microscopic or gross hematuria (cola-colored, brown, smoky, or frankly bloody urine), proteinuria (sometimes in the nephrotic range), oliguria, edema, hypertension, and acute kidney injury. Fever is unusual and suggests persistent infection.
Kidney failure, which can cause fluid overload with heart failure and severe hypertension requiring dialysis, affects approximately 1% of children. with PIGN (1).
Hematuria and hypertension may persist after disease resolution. Uncommonly, nephrotic syndrome may persist after resolution of severe disease.
Clinical manifestations of nonstreptococcal PIGN may mimic other disorders (eg, polyarteritis nodosa, renal emboli, antimicrobial drug–induced acute interstitial nephritis).
Symptoms and signs reference
1. Ong LT. Management and outcomes of acute post-streptococcal glomerulonephritis in children. World J Nephrol 2022;11(5):139-145. doi:10.5527/wjn.v11.i5.139
Diagnosis of Postinfectious Glomerulonephritis
Clinical evidence of recent infection
Urinalysis typically showing dysmorphic red blood cells (RBCs), RBC casts, proteinuria, white blood cells (WBCs), and renal tubular cells
Often hypocomplementemia
Streptococcal PIGN is suggested by history of pharyngitis or impetigo plus either typical symptoms of PIGN or incidental findings on urinalysis. Demonstration of hypocomplementemia is essentially confirmatory.
Tests performed to confirm the diagnosis depend on clinical findings. Antistreptolysin O, antihyaluronidase, and antideoxyribonuclease (anti-DNAase) antibodies are commonly measured. Serum creatinine and complement levels (C3 and CH50 [total hemolytic complement activity]) are also usually measured; however, in patients with typical clinical findings, some tests can be omitted. Sometimes other tests are performed. Biopsy can confirm the diagnosis but is rarely necessary.
Antistreptolysin O level, the most common laboratory evidence of recent streptococcal infection, increases and remains elevated for several months in most of patients with pharyngitis and in many patients with impetigo, but it is not specific.
Urinalysis typically shows proteinuria (0.5 to 2 g/m2/day); dysmorphic RBCs; WBCs; renal tubular cells; and possibly RBC, WBC, and granular casts. Random (spot) urinary protein/creatinine ratio is usually between 0.2 and 2 (normal, < 0.2) but may occasionally be in the nephrotic range (≥ 3 g/day).
Serum creatinine may rise rapidly but usually peaks below a level requiring dialysis.
C3 and CH50 levels fall during active disease and return to normal within 6 to 8 weeks in 80% of PIGN cases; C1q, C2, and C4 levels are only minimally decreased or remain normal. Cryoglobulinemia may appear and persist for several months, whereas circulating immune complexes are detectable for only a few weeks.
Biopsy specimens show enlarged and hypercellular glomeruli, initially with neutrophilic infiltration and later with mononuclear infiltration. Epithelial cell hyperplasia is a common early, transient feature. Microthrombosis may occur; if damage is severe, hemodynamic changes due to cellular proliferation and edema of the glomerulus cause oliguria, occasionally accompanied by epithelial crescents (formed within Bowman space from epithelial cell hyperplasia). Endothelial and mesangial cells multiply, and the mesangial regions often are greatly expanded by edema and contain neutrophils, dead cells, cellular debris, and subepithelial deposits of electron-dense material.

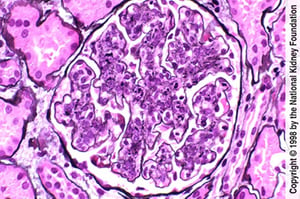
Endothelial and mesangial hypercellularity with neutrophilic infiltration (Jones silver stain, ×400).
Endothelial and mesangial hypercellularity with neutrophilic infiltration (Jones silver stain, ×400).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
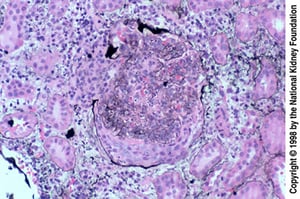
Epithelial crescents are especially common when biopsy occurs late, after a failed response to treatment. The crescent has ruptured Bowman capsule (Jones silver stain, ×200).
Epithelial crescents are especially common when biopsy occurs late, after a failed response to treatment. The crescent
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).

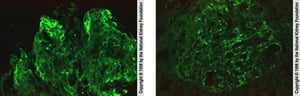
In the left image, immunofluorescent staining with anti-IgG demonstrates irregular IgG deposition in capillary loops (×400). In the right image, immunofluorescent staining with anti-C3 demonstrates scattered granular C3 deposition in capillary walls (×400).
In the left image, immunofluorescent staining with anti-IgG demonstrates irregular IgG deposition in capillary loops (×
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).

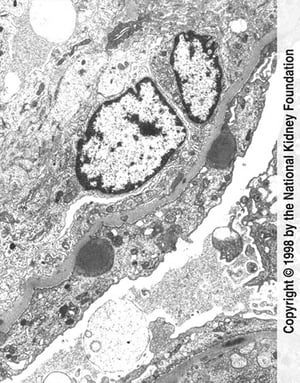
Hump-shaped immune complex deposits (dark gray) with extensive foot process effacement and endocapillary proliferation are seen on transmission electron micrograph (×11,250).
Hump-shaped immune complex deposits (dark gray) with extensive foot process effacement and endocapillary proliferation
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Endothelial and mesangial hypercellularity with neutrophilic infiltration (Jones silver stain, ×400).
Endothelial and mesangial hypercellularity with neutrophilic infiltration (Jones silver stain, ×400).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Epithelial crescents are especially common when biopsy occurs late, after a failed response to treatment. The crescent has ruptured Bowman capsule (Jones silver stain, ×200).
Epithelial crescents are especially common when biopsy occurs late, after a failed response to treatment. The crescent
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
In the left image, immunofluorescent staining with anti-IgG demonstrates irregular IgG deposition in capillary loops (×400). In the right image, immunofluorescent staining with anti-C3 demonstrates scattered granular C3 deposition in capillary walls (×400).
In the left image, immunofluorescent staining with anti-IgG demonstrates irregular IgG deposition in capillary loops (×
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Hump-shaped immune complex deposits (dark gray) with extensive foot process effacement and endocapillary proliferation are seen on transmission electron micrograph (×11,250).
Hump-shaped immune complex deposits (dark gray) with extensive foot process effacement and endocapillary proliferation
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Immunofluorescence microscopy usually shows immune complex deposition with IgG and complement in a granular pattern. On electron microscopy, these deposits are semilunar or hump-shaped and are located in the subepithelial area. The presence of these deposits and of small subendothelial and mesangial deposits initiates a complement-mediated inflammatory reaction that leads to glomerular damage. The major antigen is probably the zymogen cysteine proteinase exotoxin B (zymogen/SPE B).
Treatment of Postinfectious Glomerulonephritis
Supportive care
Treatment is supportive and may include restriction of dietary protein, sodium, and fluid and, in more severe cases, treatment of edema and hypertension. Dialysis is occasionally necessary. In clinical practice, antimicrobial therapy appears to be preventive only when given within 36 hours of infection and before glomerulonephritis becomes established.
Prognosis for Postinfectious Glomerulonephritis
Normal kidney function is retained or regained by the majority of patients. Glomerular filtration rate (GFR) usually returns to normal over 1 to 3 months, but proteinuria and microscopic hematuria may persist for up to 5 years (1). Transient changes in urinary sediment may recur with minor upper respiratory infections. Renal cellular proliferation disappears within weeks, but residual sclerosis is common. In 10% of adults and 1% of children, postinfectious glomerulonephritis evolves into rapidly progressive glomerulonephritis.
Prognosis reference
1. Satoskar AA, Parikh SV, Nadasdy T. Epidemiology, pathogenesis, treatment and outcomes of infection-associated glomerulonephritis. Nat Rev Nephrol 2020;16(1):32-50. doi:10.1038/s41581-019-0178-8
Key Points
Consider postinfectious glomerulonephritis in young patients who have had pharyngitis or impetigo plus signs of glomerulonephritis.
Antibody titers support the diagnosis and demonstration of hypocomplementemia is essentially confirmatory.
Biopsy confirms the diagnosis but is rarely necessary.
Supportive treatment usually allows recovery of kidney function.





