



Rapidly progressive glomerulonephritis is acute nephritic syndrome accompanied by microscopic glomerular crescent formation with progression to kidney failure within weeks to months. Diagnosis is based on history, urinalysis, serologic tests, and renal biopsy. Treatment is with corticosteroids, with or without cyclophosphamide or rituximab, and sometimes plasma exchange.
Rapidly progressive glomerulonephritis (RPGN), a type of nephritic syndrome, is a pathologic diagnosis accompanied by extensive glomerular crescent formation (> 50% of sampled glomeruli in a biopsy specimen contain crescents); if left untreated, RPGN progresses to kidney failure over weeks to months. It is relatively uncommon, with incidence varying by the cause of glomerulonephritis, and occurs most frequently with antiglomerular basement membrane disease (Goodpasture syndrome), antineutrophil cytoplasmic antibody (ANCA)-associated forms of glomerulonephritis, and immune complex–mediated glomerulonephritis (1). In one study of poststreptococcal glomerulonephritis, for example, approximately 12% of patients progressed to RPGN (2). Types and causes are classified by findings using immunofluorescence microscopy and serologic tests (see table ).
Classification of Rapidly Progressive Glomerulonephritis Based on Immunofluorescence Microscopy
Type | Approximate Percentage of RPGN Cases* | Causes |
|---|---|---|
Anti-GBM antibody–mediated | 20% | Anti-GBM GN (without lung hemorrhage†) Goodpasture syndrome (with lung hemorrhage) |
Immune complex-mediated | 50% | Postinfectious causes:
Systemic rheumatic diseases:
Other glomerulopathies:
|
Pauci-immune | 30% | Granulomatosis with polyangiitis Renal-limited vasculitis (eg, idiopathic crescentic GN) |
Double-antibody positive | Rare | Same as for anti-GBM antibody-mediated and pauci-immune types |
* Data from Satoskar AA, Parikh SV, Nadasdy T. Epidemiology, pathogenesis, treatment and outcomes of infection-associated glomerulonephritis. Nat Rev Nephrol 2020;16(1):32-50. doi:10.1038/s41581-019-0178-8 | ||
† When the lung is also affected, anti-GBM glomerulonephritis is called Goodpasture syndrome. | ||
GBM = glomerular basement membrane; GN = glomerulonephritis; RPGN = rapidly progressive glomerulonephritis; IgA = immunoglobulin A. | ||
Antiglomerular basement membrane antibody disease
Antiglomerular basement membrane (GBM) antibody disease is an autoimmune glomerulonephritis and an important cause of RPGN. It may arise when respiratory exposures (eg, cigarette smoke, viral upper respiratory infection) or some other stimulus exposes alveolar capillary collagen, triggering formation of anticollagen antibodies. The anticollagen antibodies cross-react with GBM, fixing complement and triggering a cell-mediated inflammatory response in the kidneys and usually the lungs.
The term Goodpasture syndrome refers to a combination of glomerulonephritis and alveolar hemorrhage in the presence of anti-GBM antibodies. Glomerulonephritis without alveolar hemorrhage in the presence of anti-GBM antibodies is called anti-GBM glomerulonephritis. Immunofluorescent staining of renal biopsy tissue demonstrates linear IgG deposits.
Immune complex RPGN
Immune complex RPGN complicates numerous infectious and systemic rheumatic diseases and also occurs with other primary glomerulopathies.
Immunofluorescent staining demonstrates nonspecific granular immune deposits. The condition accounts for up to 40% of RPGN cases. Pathogenesis is usually unknown.
Pauci-immune RPGN
Pauci-immune RPGN is distinguished by the absence of immune complex or complement deposition on immunofluorescent staining. It constitutes the majority of all RPGN cases (3). Almost all patients have elevated antineutrophil cytoplasmic antibodies (ANCAs), usually antiproteinase 3-ANCA or myeloperoxidase-ANCA, and systemic vasculitis.
Double-antibody positive disease
Double-antibody positive disease occurs with the presence of anti-GBM and ANCA antibodies. It is rare.
Idiopathic RPGN
Idiopathic cases are rare. They include patients with either of the following:
Immune complexes but no obvious cause such as infection, systemic rheumatic disease, or glomerular disorder
Pauci-immune features but absence of ANCA antibodies
General references
1. Sethi S, De Vriese AS, Fervenza FC. Acute glomerulonephritis. Lancet 2022;399(10335):1646-1663. doi:10.1016/S0140-6736(22)00461-5
2. Karakaya D, Güngör T, Çakıcı EK, et al. Predictors of rapidly progressive glomerulonephritis in acute poststreptococcal glomerulonephritis. Pediatr Nephrol 2023;38(9):3027-3033. doi:10.1007/s00467-023-05935-9
3. Syed R, Rehman A, Valecha G, El-Sayegh S. Pauci-Immune Crescentic Glomerulonephritis: An ANCA-Associated Vasculitis. Biomed Res Int 2015;2015:402826. doi:10.1155/2015/402826
Symptoms and Signs of RPGN
Manifestations are usually insidious, with weakness, fatigue, fever, nausea, vomiting, anorexia, arthralgia, and abdominal pain. Some patients present similarly to those with postinfectious glomerulonephritis, with abrupt-onset hematuria. About half of patients have edema and a history of an acute influenza-like illness within 4 weeks of onset of kidney failure, usually followed by severe oliguria. Nephrotic syndrome may be present. Hypertension is uncommon and rarely severe. Patients with anti-GBM antibody disease may have pulmonary hemorrhage, which can manifest with hemoptysis or be detectable only by findings suggestive of diffuse alveolar infiltrates on lung imaging (pulmonary-renal syndrome or diffuse alveolar hemorrhage syndrome).
Diagnosis of RPGN
Progressive kidney failure over weeks to months
Nephritic urinary sediment
Serologic testing
Serum complement levels
Renal biopsy
Diagnosis is suggested by acute kidney injury in patients with hematuria and dysmorphic red blood cells (RBCs) or RBC casts. Testing includes serum creatinine, urinalysis, complete blood count (CBC), serologic tests, and renal biopsy. Diagnosis is usually by serologic tests and renal biopsy.
Serum creatinine is almost always elevated.
Urinalysis shows hematuria is always present, and RBC casts are usually present. Telescopic sediment (ie, sediment with multiple elements, including white blood cells [WBCs]; dysmorphic RBCs; and WBC, RBC, granular, waxy, and broad casts) is common.
On CBC, anemia is usually present, and leukocytosis is common.
Serologic testing should include anti-GBM antibodies (anti-GBM antibody disease); antistreptolysin O antibodies, anti-DNA antibodies, or cryoglobulins (immune complex RPGN); and antineutrophil cytoplasmic antibodies (ANCA) titers (pauci-immune RPGN).
Complement measurement (serum C3 and C4) may be useful in suspected immune complex RPGN because hypocomplementemia is common.
Early renal biopsy is essential. The feature common to all types of RPGN is focal proliferation of glomerular epithelial cells, sometimes interspersed with numerous neutrophils, that forms a crescentic cellular mass (crescents) and that fills Bowman space in > 50% of glomeruli. The glomerular tuft usually appears hypocellular and collapses. Necrosis within the tuft or involving the crescent may occur and may be the most prominent abnormality. In such patients, histologic evidence of vasculitis should be sought.

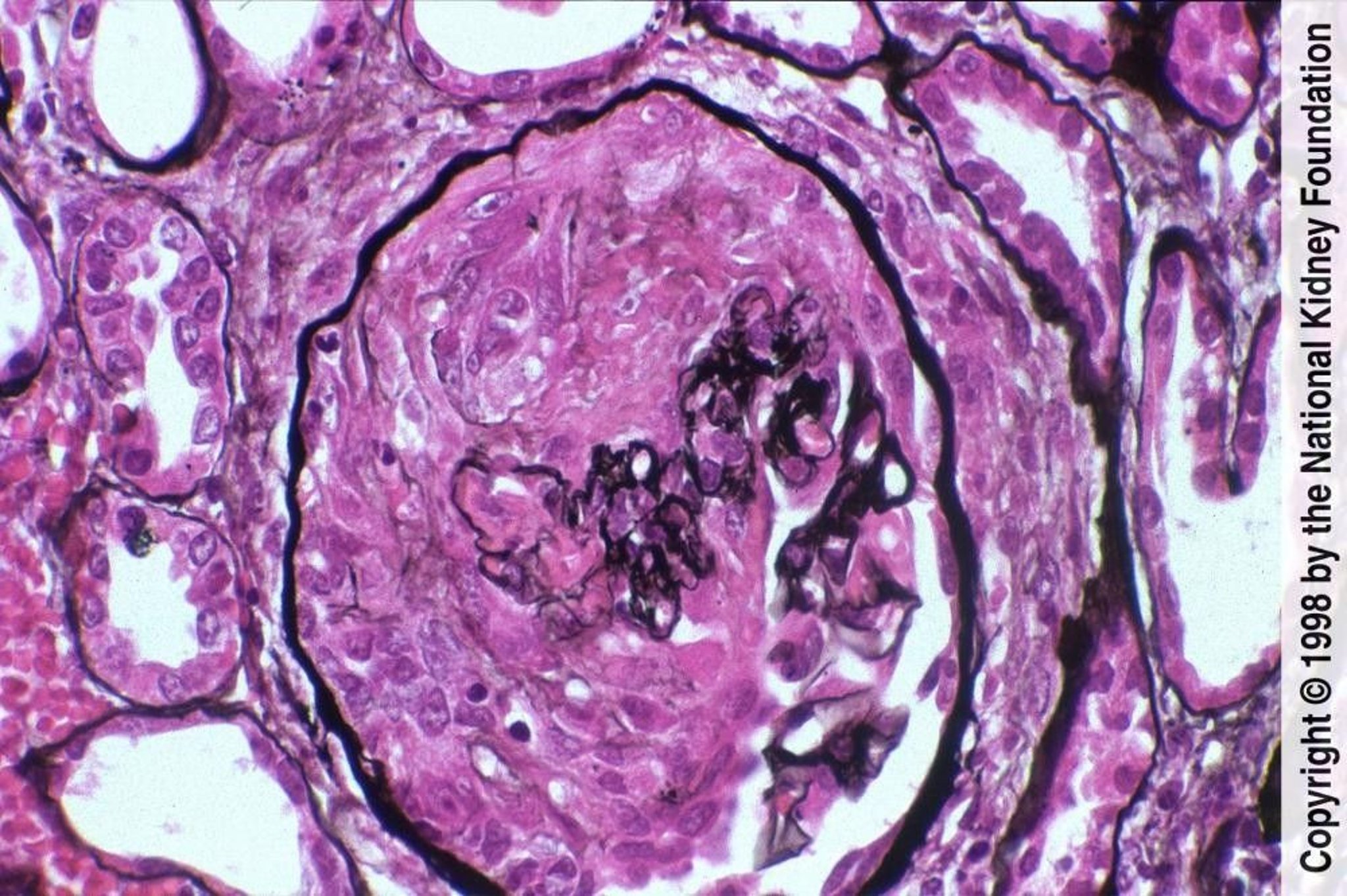
Pauci-immune disease is characterized by crescent formation with fibrinoid necrosis and negative immunofluorescence staining (Jones silver stain, ×400).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
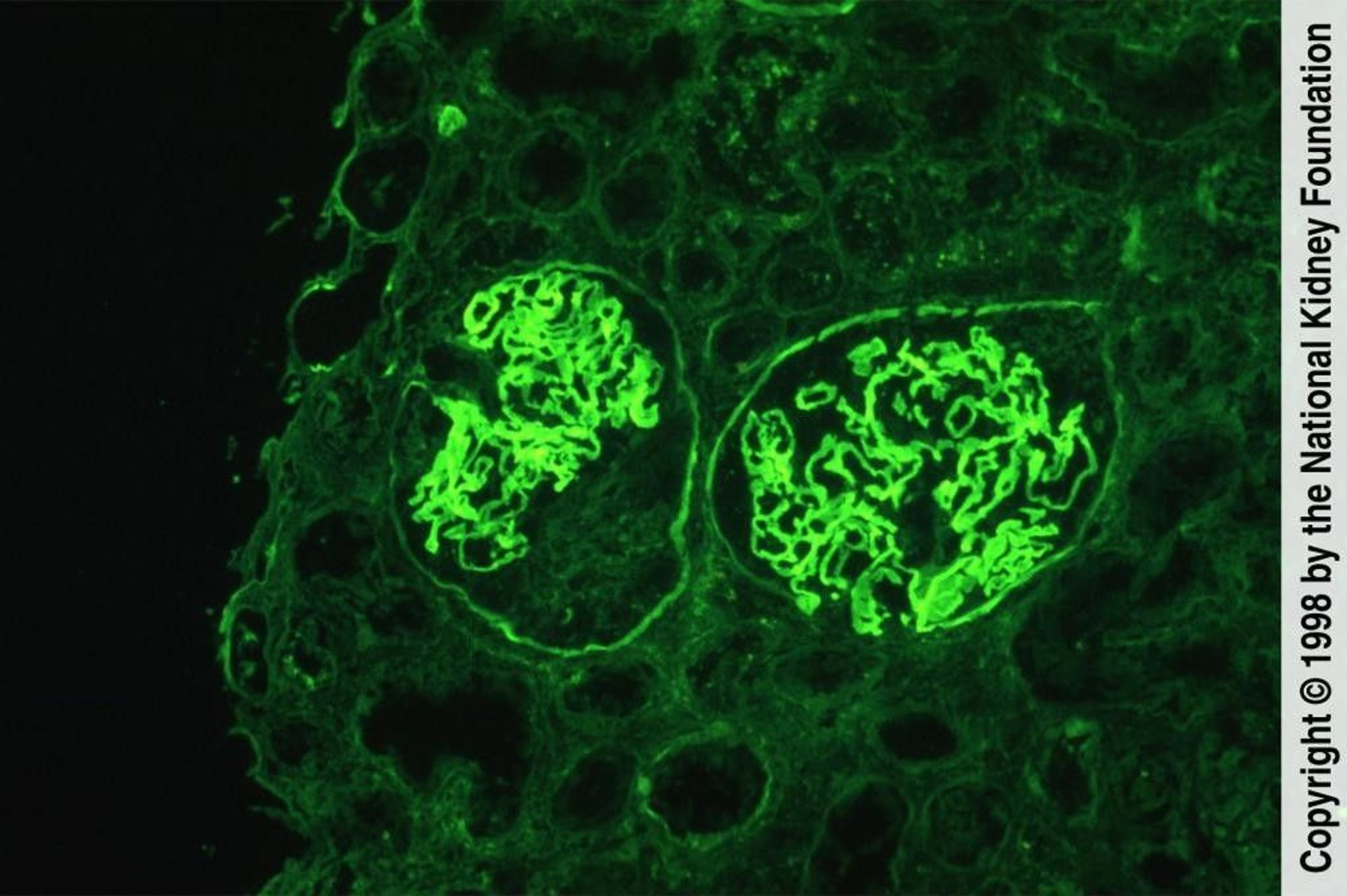
Antiglomerular basement membrane antibody disease is characterized by smooth, linear staining of glomerular basement membranes with antibody to IgG. The left glomerulus demonstrates crescent formation (immunofluorescence with anti-IgG, ×200).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Immunofluorescence microscopy findings differ for each type:
In anti-GBM antibody disease, linear or ribbon-like deposition of IgG along the GBM is most prominent and is often accompanied by linear and sometimes granular deposition of C3.
In immune complex RPGN, immunofluorescence reveals diffuse, irregular mesangial IgG and C3 deposits.
In pauci-immune RPGN, immune staining and deposits are not detected. However, fibrin occurs within the crescents, regardless of the fluorescence pattern.
In double antibody positive RPGN, linear staining of the GBM is present.
In idiopathic RPGN, some patients have immune complexes and others have absence of immune staining and deposits.
Treatment of RPGN
Corticosteroids
Cyclophosphamide
Rituximab
Plasma exchange
Treatment varies by disease type, although no regimens have been rigorously studied. Therapy should be instituted early, ideally when serum creatinine is < 5 mg/dL (442 micromol/L) and before the biopsy shows crescentic involvement of all glomeruli or organizing crescents as well as fibrotic interstitium and atrophic tubules. Even patients with kidney involvement and higher creatinine levels should be aggressively treated if they do not require immediate kidney replacement therapy. Treatment becomes less effective as these features become more prominent and may be harmful in some patients (eg, older patients, patients with infection).
Corticosteroids and either cyclophosphamide or rituximab are usually given. For immune complex and pauci-immune disease, corticosteroids (methylprednisolone 1 g IV once a day over 30 minutes for 3 to 5 days followed by prednisone 1 mg/kg orally once a day) may reduce serum creatinine levels or delay dialysis for > 3 years in 50% of patients (1, 2).
Cyclophosphamide is usually given and may particularly benefit antineutrophil cytoplasmic antibody (ANCA)–positive patients; IV monthly pulse regimens may cause fewer adverse effects (eg, leukopenia, infection) than oral therapy because of reduced cumulative dosing. Prednisone and cyclophosphamide are typically started concurrent with plasma exchange for anti-GBM (glomerular basement membrane) antibody disease and continued to minimize new antibody formation. Patients with idiopathic disease are usually treated with corticosteroids and cyclophosphamide, but data regarding efficacy are scarce.
Rituximab may be dosed at 375 mg/m2 IV per week for 4 weeks as used in the RAVE trial (formal title: Rituximab in ANCA-Associated Vasculitis; [2]). An alternative regimen is an initial dose of 1g followed by another 1-g dose 2 weeks later.
Plasma exchange (for 14 days) is recommended for anti-GBM antibody disease. Plasma exchange may also be considered for immune complex and pauci-immune ANCA-associated RPGN with pulmonary hemorrhage or severe kidney disease on presentation (serum creatinine > 5 to 7 mg/dL [442 to 618.8 micromol/L] or dialysis dependency), but its use remains controversial. Plasma exchange is thought to rapidly remove free antibody, intact immune complexes, and mediators of inflammation (eg, fibrinogen, complement). While some evidence suggested that plasma exchange improved short-term kidney outcomes, a subsequent randomized trial did not show that it reduced the incidence of death or kidney failure (3).
Aggressive immunosuppressive therapy may also be beneficial in patients who present with higher creatinine levels for example, plasmapheresis combined with prednisone and cyclophosphamide benefited patients with kidney involvement who did not require immediate kidney replacement therapy, even if creatinine levels were elevated above 5 to 7 mg/dL (442 to 618.8 micromol/L [4]).
Kidney transplantation is effective for all types, but disease may recur in the graft; risk diminishes with time. In anti-GBM antibody disease, the anti-GBM titers should be undetectable for at least 12 months before transplantation. For patients with pauci-immune RPGN, disease activity should be quiescent for at least 6 months before transplantation; ANCA titers do not need to be suppressed.
Treatment references
1. Ponticelli C, Altieri P, et al: A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol 9(3):444, 1998. doi: 10.1681/ASN.V93444
2. Jones RB, Cohen Tervaert JW, Hauser T: Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 363:211-220, 2010. doi: 10.1056/NEJMoa0909169
3. Walsh M, Merkel PA, Peh C-A, et al: Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med 382(7):621-631, 2020. doi: 10.1056/NEJMoa1803537
4. Levy JB, Turner AN, Rees AJ, et al: Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med 134(11):1033-1042, 2001. doi: 10.7326/0003-4819-134-11-200106050-00009
Prognosis for RPGN
Spontaneous remission is rare, and up to 90% of untreated patients progress to kidney failure (1). Prognosis improves with early treatment.
Favorable prognostic factors include RPGN caused by the following:
Anti-GBM disease if treated early, especially when treated before oliguria occurs and when creatinine level is < 7 mg/dL (618.8 micromol/L)
Unfavorable prognostic factors include the following:
Age > 60 years
Oliguric kidney failure
Higher serum creatinine level
Circumferential crescents in > 75% of glomeruli
Pauci-immune RPGN
Up to half of patients with pauci-immune RPGN respond to treatment; among patients who respond to treatment, < 10% of patients require dialysis and approximately 15% die from the disease within 5 years; in contrast, among nonresponders, more than 50% require dialysis and approximately 40% die within 5 years (2).
Patients with double-antibody disease appear to have a renal prognosis somewhat better than patients with only anti-GBM antibody disease and worse than patients with pauci-immune disease.
Patients who recover normal kidney function after RPGN demonstrate residual histologic changes principally in glomeruli, consisting chiefly of hypercellularity, with little or no sclerosis within the glomerular tuft or the epithelial cells and minimal fibrosis of the interstitium.
Death is usually due to infectious or cardiac causes, providing that a uremic death is prevented by dialysis.
Prognosis references
1. Zäuner I, Bach D, Braun N, et al. Predictive value of initial histology and effect of plasmapheresis on long-term prognosis of rapidly progressive glomerulonephritis. Am J Kidney Dis 2002;39(1):28-35. doi:10.1053/ajkd.2002.29874
2. Lee T, Gasim A, Derebail VK, et al. Predictors of treatment outcomes in ANCA-associated vasculitis with severe kidney failure. Clin J Am Soc Nephrol 2014;9(5):905-913. doi:10.2215/CJN.08290813
Key Points
Consider rapidly progressive glomerulonephritis if patients have acute kidney injury with hematuria and dysmorphic red blood cells (RBCs) or RBC casts, particularly with subacute constitutional or nonspecific symptoms (eg, fatigue, fever, anorexia, arthralgia, abdominal pain).
Evaluate with serologic tests and early renal biopsy.
Initiate treatment early, with corticosteroids, cyclophosphamide, and in some cases plasma exchange.
Consider kidney transplantation after disease activity is controlled.
Drug Information for the Topic





