



11Beta-hydroxylase (CYP11B1) deficiency involves defective production of cortisol, with accumulation of mineralocorticoid precursors, resulting in hypernatremia, hypokalemia, and hypertension and increased production of adrenal androgens, leading to virilization. Diagnosis is by measurement of cortisol, its precursors, and adrenal androgens and sometimes by measuring 11-deoxycortisol after adrenocorticotropic hormone administration. Treatment is with a glucocorticoid.
11Beta-hydroxylase deficiency, the second most common cause of congenital adrenal hyperplasia, is responsible for up to 8% of all cases of congenital adrenal hyperplasia (1, 2, 3, 4). It is caused by mutations in the CYP11B1 gene (5). Conversion of 11-deoxycortisol to cortisol and deoxycorticosterone to corticosterone is partially blocked, leading to:
Increased levels of adrenocorticotropic hormone (ACTH)
Accumulation of 11-deoxycortisol (which has limited biological activity) and deoxycorticosterone (which has mineralocorticoid activity)
Overproduction of adrenal androgens (dehydroepiandrosterone [DHEA], androstenedione, and testosterone)
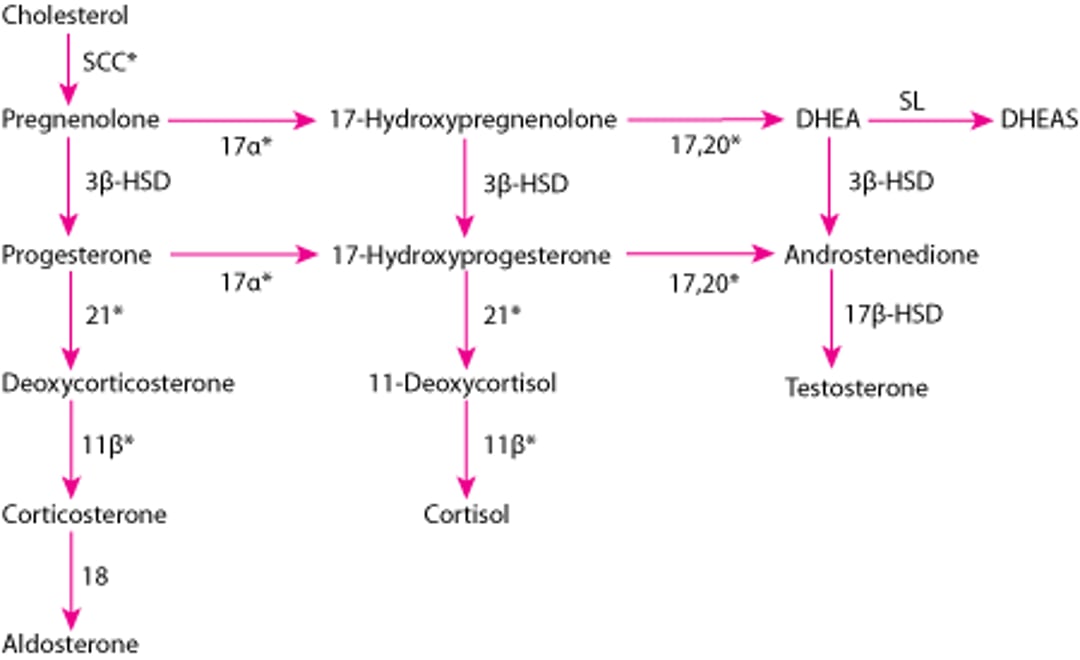
Adrenal Hormone Synthesis
* Enzymes stimulated by adrenocorticotropic hormone (ACTH). |
11β = 11β-hydroxylase (P-450c11); 17α = 17α-hydroxylase (P-450c17); 17,20 = 17,20 lyase (P-450c17); 18 = aldosterone synthase (P-450aldo); 21 = 21-hydroxylase (P-450c21); DHEA = dehydroepiandrosterone; DHEAS = DHEA sulfate; 3β-HSD = 3β-hydroxysteroid dehydrogenase (3β2-HSD); 17β-HSD = 17β-hydroxysteroid dehydrogenase (17β-HSD); SCC = side-chain cleavage (P-450scc); SL = sulfotransferase (SULT1A1, SULT1E1). |

General references
1. Witchel SF: Congenital adrenal hyperplasia. J Pediatr Adolesc Gynecol 30(5):520–534, 2017. doi: 10.1016/j.jpag.2017.04.001
2. El-Maouche D, Arlt W, Merke DP: Congenital adrenal hyperplasia. Lancet 390(10108):2194–2210, 2017. doi: 10.1016/S0140-6736(17)31431-9
3. Breil T, Yakovenko V, Inta I, et al. Typical characteristics of children with congenital adrenal hyperplasia due to 11β-hydroxylase deficiency: a single-centre experience and review of the literature. J Pediatr Endocrinol Metab. 2019;32(3):259-267. doi:10.1515/jpem-2018-0298
4. Bulsari K, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Endocrine. 2017;55(1):19-36. doi:10.1007/s12020-016-1189-x
5. Auer MK, Nordenström A, Lajic S, Reisch N. Congenital adrenal hyperplasia. Lancet. 2023;401(10372):227-244. doi:10.1016/S0140-6736(22)01330-7
Symptoms and Signs of 11Beta-Hydroxylase Deficiency
Female neonates may present with genital ambiguity, including clitoral enlargement, labial fusion, and a urogenital sinus. Male neonates usually appear normal, but some present with penile enlargement (see also Adrenal Virilism).
Males and females may present later with precocious puberty. Females with milder disease (with higher residual enzyme activity) may have normal genitalia and may present later with menstrual irregularities, acne, and hirsutism.
Salt retention with hypernatremia, hypertension, and hypokalemic alkalosis may result from increased mineralocorticoid activity due to increased deoxycorticosterone levels.
Diagnosis of 11Beta-Hydroxylase Deficiency
Plasma levels of 11-deoxycortisol and adrenal androgens
Prenatal diagnosis is available though rarely performed (1). Diagnosis of 11beta-hydroxylase deficiency in neonates is established by increased plasma levels of 11-deoxycortisol and adrenal androgens (DHEA, androstenedione, and testosterone). Plasma renin activity is often suppressed because of increased mineralocorticoid activity; measuring plasma renin activity may be useful in older children but is less reliable in neonates. If the diagnosis is uncertain, levels of 11-deoxycortisol and adrenal androgens are measured before and 60 minutes after ACTH stimulation (2). In affected adolescents, basal plasma levels of 11-deoxycortisol and adrenal androgens may be normal, so ACTH stimulation should be used.
Hypertension occurs in about two-thirds of patients with 11beta-hydroxylase deficiency (caused by CYP11B1 mutation) and distinguishes it from 21-hydroxylase deficiency (usually caused by CYP21A2 mutation), which causes hypotension. Because both 11beta-hydroxylase deficiency and 21-hydroxylase deficiency can cause increased levels of 17-hydroxyprogesterone, which is measured during routine newborn screening, patients with mild to moderately increased levels of 17-hydroxyprogesterone should have 11-deoxycortisol levels measured. Hypokalemia may occur but not in all patients.
Pearls & Pitfalls
|
Diagnosis references
1. Motaghedi R, Betensky BP, Slowinska B, et al. Update on the prenatal diagnosis and treatment of congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. J Pediatr Endocrinol Metab. 2005;18(2):133-142. doi:10.1515/jpem.2005.18.2.133
2. Bulsari K, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Endocrine. 2017;55(1):19-36. doi:10.1007/s12020-016-1189-x
Treatment of 11Beta-Hydroxylase Deficiency
Glucocorticoid replacement
Sometimes antihypertensive therapy
Treatment of 11beta-hydroxylase deficiency is cortisol replacement, typically with hydrocortisone orally 2 to 3 times a day, which prevents further virilization and ameliorates hypertension by reducing levels of 11-deoxycortisol, deoxycorticosterone, and adrenal androgens that are stimulated by ACTH. Unlike the salt-wasting form of 21-hydroxylase deficiency, mineralocorticoid replacement is not required, because sodium and potassium homeostasis is maintained as a result of mineralocorticoid effects of deoxycorticosterone.
Response to treatment should be monitored, typically by measuring serum 11-deoxycortisol and adrenal androgens and by assessing growth velocity and skeletal maturation. Blood pressure should be monitored closely in patients who presented with hypertension. Antihypertensives, such as potassium-sparing diuretics or calcium channel blockers, may be required (1).
Treatment reference
1. Bulsari K, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Endocrine. 2017;55(1):19-36. doi:10.1007/s12020-016-1189-x
Key Points
Children with 11beta-hydroxylase deficiency have excess mineralocorticoid activity and increased adrenal androgens, which cause hypertension, hypokalemia, and virilization.
In females, androgen excess usually manifests at birth with ambiguous external genitals (eg, clitoral enlargement, fusion of the labia majora, a urogenital sinus rather than distinct urethral and vaginal openings); later in life they may have precocious puberty or hirsutism, oligomenorrhea, and acne.
Male infants usually appear normal but may later have precocious puberty.
Diagnose by steroid hormone levels often after adrenocorticotropic hormone stimulation.
Treat with glucocorticoid replacement and sometimes antihypertensives.
Drug Information for the Topic





