



21-Hydroxylase (CYP21A2) deficiency causes defective conversion of adrenal precursors to cortisol and, in some cases, to aldosterone, sometimes resulting in severe hyponatremia and hyperkalemia. Accumulated hormone precursors are shunted into androgen production, causing virilization. Diagnosis is by measurement of cortisol, its precursors, and adrenal androgens, sometimes after adrenocorticotropic hormone administration. Treatment is with a glucocorticoid plus, if needed, a mineralocorticoid.
21-Hydroxylase deficiency causes 95% of all cases of congenital adrenal hyperplasia (1).
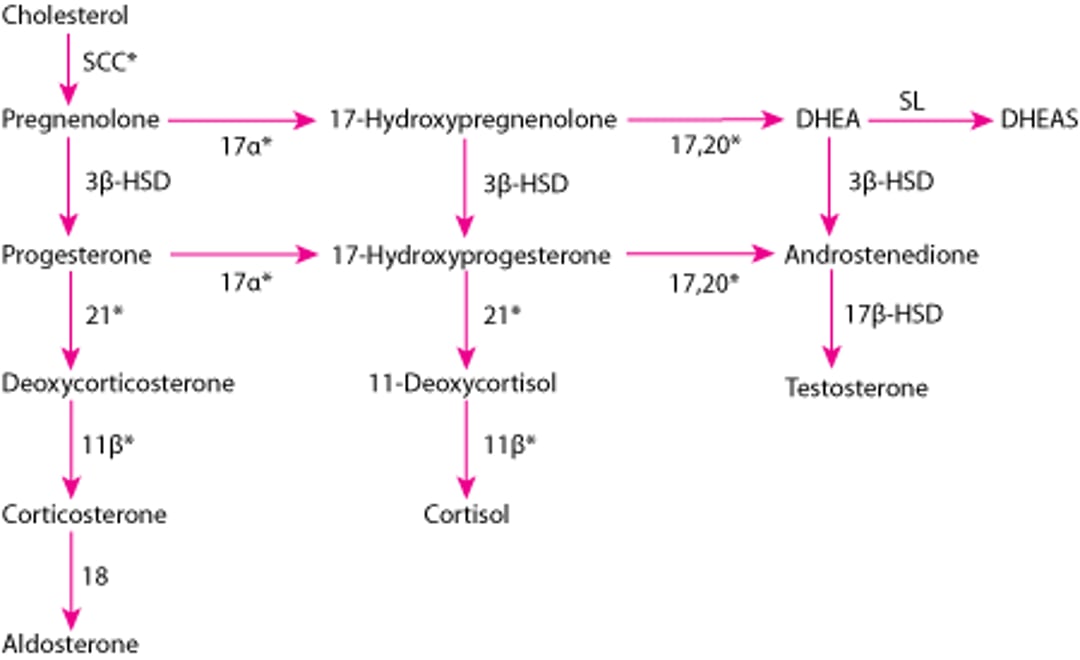
Disease phenotype and severity depend on the specific CYP21A2 mutation and degree of enzyme deficiency (1, 2). The deficiency completely or partially blocks conversion of 17-hydroxyprogesterone to 11-deoxycortisol, a precursor of cortisol, and conversion of progesterone to deoxycorticosterone, a precursor of aldosterone. Because cortisol synthesis is decreased, adrenocorticotropic hormone (ACTH) levels increase, which stimulates the adrenal cortex, causing accumulation of cortisol precursors (eg, 17-hydroxyprogesterone) and excessive production of the adrenal androgens dehydroepiandrosterone (DHEA) and androstenedione. Aldosterone deficiency can lead to salt wasting, hyponatremia, and hyperkalemia.
Adrenal Hormone Synthesis
* Enzymes stimulated by adrenocorticotropic hormone (ACTH). |
11β = 11β-hydroxylase (P-450c11); 17α = 17α-hydroxylase (P-450c17); 17,20 = 17,20 lyase (P-450c17); 18 = aldosterone synthase (P-450aldo); 21 = 21-hydroxylase (P-450c21); DHEA = dehydroepiandrosterone; DHEAS = DHEA sulfate; 3β-HSD = 3β-hydroxysteroid dehydrogenase (3β2-HSD); 17β-HSD = 17β-hydroxysteroid dehydrogenase (17β-HSD); SCC = side-chain cleavage (P-450scc); SL = sulfotransferase (SULT1A1, SULT1E1). |

Classic (severe) 21-hydroxylase deficiency
Classic 21-hydroxylase deficiency occurs in 1/10,000 to 1/20,000 live births, as diagnosed by newborn screening (2), and can be divided into 2 forms:
Salt wasting
Simple virilizing
In both forms, adrenal androgen levels are elevated, causing virilization.
The salt-wasting form is the more severe and accounts for the majority of classic 21-hydroxylase deficiency cases (75% of infants with classic 21-hydroxylase deficiency experience a salt-wasting adrenal crisis) (2); there is complete (< 1% of normal activity) deficiency of enzyme activity that leads to very low levels of cortisol and aldosterone. Because minimal aldosterone is secreted, salt is lost, leading to hyponatremia, hyperkalemia, and increased plasma renin activity.
In the simple virilizing form, cortisol synthesis is impaired, leading to increased androgen activity, but there is sufficient enzyme activity (1 to 10% of normal activity) to maintain normal, or only slightly decreased, aldosterone production(1).
Nonclassic (mild) 21-hydroxylase deficiency
Nonclassic 21-hydroxylase deficiency is more common than classic 21-hydroxylase deficiency. Incidence ranges from approximately 1/1000live births in White people (0.1%) to approximately 3% in certain ethnic groups (eg, people of Ashkenazi Jewish ancestry) (3).
Nonclassic 21-hydroxylase deficiency causes a less severe form of the disorder in which there is 20 to 80% of normal 21-hydroxylase activity(1). Salt wasting is absent because aldosterone and cortisol levels are normal; however, adrenal androgen levels are slightly elevated, resulting in mild androgen excess in childhood or adulthood.
General references
1. Auer MK, Nordenström A, Lajic S, Reisch N. Congenital adrenal hyperplasia. Lancet. 2023;401(10372):227-244. doi:10.1016/S0140-6736(22)01330-7
2. Merke DP, Auchus RJ. Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. N Engl J Med. 2020;383(13):1248-1261. doi:10.1056/NEJMra1909786
3. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. 2015;50(1):32-50. doi:10.1007/s12020-015-0656-0
Symptoms and Signs of 21-Hydroxylase Deficiency
The salt-wasting form of classic 21-hydroxylase deficiency causes hyponatremia (sometimes severe), hyperkalemia, and hypotension as well as virilization. If undiagnosed and untreated, this form can lead to life-threatening adrenal crisis, with vomiting, diarrhea, hypoglycemia, hypovolemia, and shock.
With either form of classic 21-hydroxylase deficiency, female neonates have ambiguous external genitals, with clitoral enlargement, fusion of the labia majora, and a urogenital sinus rather than distinct urethral and vaginal openings. Male infants typically have normal genital development, which can delay the diagnosis of the salt-wasting form; affected boys are often identified only through routine newborn screening.
Unless detected by newborn screening, boys with the simple virilizing form may not be diagnosed for several years, when they develop signs of androgen excess. Signs of androgen excess may include early appearance of pubic hair and increase in growth velocity in both sexes, clitoral enlargement in girls, and penile enlargement and earlier deepening of voice in boys.
In affected females, especially those with the salt-wasting form, reproductive function may be impaired as they reach adulthood; they may have labial fusion and anovulatory cycles or amenorrhea. Some males with the salt-wasting form are fertile as adults, but others may develop testicular adrenal rest tumors (benign intratesticular masses composed of adrenal tissue that hypertrophies under chronic ACTH stimulation), Leydig cell dysfunction, decreased testosterone, and impaired spermatogenesis. Males with classic 21-hydroxylase deficiency, even if untreated, are often (or, according to some studies, usually) fertile, but spermatogenesis is often impaired (2, 3).
Children with nonclassic 21-hydroxylase deficiency do not have symptoms at birth and usually do not present until childhood or adolescence (1). Affected females may have early pubic hair development, advanced bone age, hirsutism, oligomenorrhea, and/or acne; these symptoms may resemble the manifestations of polyendocrine metabolic ovarian syndrome. Affected males may have early pubic hair development, growth acceleration, and advanced bone age.
Symptoms and signs references
1. Auer MK, Nordenström A, Lajic S, Reisch N. Congenital adrenal hyperplasia. Lancet. 2023;401(10372):227-244. doi:10.1016/S0140-6736(22)01330-7
2. Bouvattier C, Esterle L, Renoult-Pierre P, et al. Clinical Outcome, Hormonal Status, Gonadotrope Axis, and Testicular Function in 219 Adult Men Born With Classic 21-Hydroxylase Deficiency. A French National Survey. J Clin Endocrinol Metab. 2015;100(6):2303-2313. doi:10.1210/jc.2014-4124
3. King TF, Lee MC, Williamson EE, Conway GS. Experience in optimizing fertility outcomes in men with congenital adrenal hyperplasia due to 21 hydroxylase deficiency. Clin Endocrinol (Oxf). 2016;84(6):830-836. doi:10.1111/cen.13001
Diagnosis of 21-Hydroxylase Deficiency
Newborn screening
Additional blood tests
Possibly ACTH stimulation test
Possibly genotyping (including prenatal genetic testing)
Routine newborn screening typically includes measuring serum levels of 17-hydroxyprogesterone. If levels are elevated, the diagnosis of 21-hydroxylase deficiency is confirmed by identifying low blood levels of cortisol and by identifying high blood levels of DHEA, androstenedione, and testosterone. Rarely, the diagnosis is uncertain, and levels of these hormones must be measured before and 60 minutes after ACTH is given (ACTH or cosyntropin stimulation test). Genetic testing is performed if results are inconclusive (1). In patients who develop symptoms later, ACTH stimulation testing may help, but genotyping may be required.
Children with the salt-wasting form have hyponatremia and hyperkalemia; low levels of deoxycorticosterone, corticosterone, and aldosterone; and high levels of renin.
Prenatal screening and diagnosis (and experimental treatment) are possible; CYP21 genes are analyzed if risk is high (eg, the fetus has an affected sibling with the genetic defect). Carrier status (heterozygosity) can be determined in children and adults.
Diagnosis reference
1. Speiser PW, Arlt W, Auchus RJ, et al: Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103(11):4043-4088. doi: 10.1210/jc.2018-01865
Treatment of 21-Hydroxylase Deficiency
Glucocorticoid replacement
Mineralocorticoid replacement (salt-wasting form)
Adrenal crisis in infants may result in hypotension and shock, accompanied by hyponatremia and hyperkalemia. Depending on age, it may manifest with lethargy, fatigue, vomiting, confusion, or even coma. Urgent therapy with IV fluids and glucocorticoids is needed. Stress doses of hydrocortisone (100 mg/m2/day) are given initially intravenously if the salt-wasting form is suspected with an initial bolus of 100 mg/m2/day hydrocortisone, typically 20 to 25 mg based on infant body surface area, followed by 100 mg/m2/day intravenous dose divided every 6 hours until clinically stable; doses can be transitioned to enteral as the child stabilizes, and the dose is reduced over several weeks to a more physiologic replacement dose.
Maintenance treatment of classic 21-hydroxylase deficiency is glucocorticoids as replacement for deficient steroids (typically, a tablet form of oral hydrocortisone 3 times a day). For infants and younger children, tablets are crushed or split and mixed with liquid. Low-dose hydrocortisone granules (sprinkles) are available for the treatment of infants with congenital adrenal hyperplasia and may improve the accuracy of dosing.
Hydrocortisone is preferred in growing children because it is less potent than other glucocorticoid preparations and causes less growth suppression (1). Postpubertal adolescents and adults may be treated with prednisone orally 1 or 2 times a day, prednisolone orally 1 or 2 times a day, or dexamethasone orally 1 or 2 times a day.
Response to therapy is monitored in infants every 3 months and in children aged > 12 months every 3 to 4 months. Overtreatment with a glucocorticoid results in iatrogenic Cushing syndrome, causing obesity, subnormal growth, and delayed skeletal maturation. Undertreatment results in inability to suppress ACTH with consequent hyperandrogenism, causing virilization and supranormal growth velocity in children and, eventually, premature termination of growth and short stature. Monitoring involves measuring serum 17-hydroxyprogesterone, androstenedione, and testosterone levels as well as assessing growth velocity and skeletal maturation each year.
For the salt-wasting form, in addition to glucocorticoids, maintenance treatment includes mineralocorticoid replacement for restoration of sodium and potassium homeostasis. Oral fludrocortisone is given if salt loss occurs. Infants often require supplemental oral salt for approximately 1 year. Close monitoring during therapy is critical, especially during the first 2 years of life when salt and higher doses of fludrocortisone are needed (2). In adults and children over 4 years, crinecerfont, a corticotropin releasing factor-1 receptor antagonist, can be used as adjunctive treatment in classic congenital adrenal hyperplasia (3, 4, 5, 6). It can reduce the glucocorticoid requirement by reducing ACTH and adrenal androgen levels.
With illness, glucocorticoid dosages are increased (typically doubled or tripled) to prevent adrenal crisis. Mineralocorticoid replacement is not adjusted. When oral therapy is unreliable (eg, severe vomiting or life-threatening situations), a single IM injection of hydrocortisone can be given. When the injection is given, children typically need to be evaluated in the emergency department to determine whether they require IV fluids, additional glucocorticoids, or both.
For prenatal treatment, a glucocorticoid (usually dexamethasone) is given to the mother to suppress fetal pituitary secretion of ACTH and thus reduce or prevent virilization of affected female fetuses. Treatment, which is experimental, must begin in the first several weeks of gestation (1).
Treatment of nonclassic 21-hydroxylase deficiency depends on symptoms. If asymptomatic, no treatment is required. If symptomatic (children with early or rapid progression of pubarche or bone age, adolescents with virilization, adult women with hyperandrogenism and infertility), glucocorticoid treatment is similar to classic 21-hydroxylase deficiency (1), but lower doses are often effective. Mineralocorticoid replacement is not needed.

Treatment references
1. Speiser PW, Arlt W, Auchus RJ, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103(11):4043-4088. doi:10.1210/jc.2018-01865
2. Neumann U, van der Linde A, Krone RE, et al: Treatment of congenital adrenal hyperplasia in children aged 0-3 years: a retrospective multicenter analysis of salt supplementation, glucocorticoid and mineralocorticoid medication, growth and blood pressure. Eur J Endocrinol 186(5):587-596, 2022. doi: 10.1530/EJE-21-1085
3. Auchus RJ, Hamidi O, Pivonello R, et al. Phase 3 Trial of Crinecerfont in Adult Congenital Adrenal Hyperplasia. N Engl J Med. 2024;391(6):504-514. doi:10.1056/NEJMoa2404656
4. Sarafoglou K, Kim MS, Lodish M, et al. Phase 3 Trial of Crinecerfont in Pediatric Congenital Adrenal Hyperplasia. N Engl J Med. 2024;391(6):493-503. doi:10.1056/NEJMoa2404655
5. Yuan L. Crinecerfont: CRF1R Antagonist Approved for Treatment of Congenital Adrenal Hyperplasia. Ann Pharmacother. 2026;60(5):512-520. doi:10.1177/10600280251396594
6. Auer MK, Nordenström A, Lajic S, Reisch N. Congenital adrenal hyperplasia. Lancet. 2023;401(10372):227-244. doi:10.1016/S0140-6736(22)01330-7
Key Points
Children with 21-hydroxylase deficiency have varying degrees of androgen excess and at least 75% have a salt-wasting form caused by aldosterone deficiency.
In females, androgen excess usually manifests at birth with ambiguous external genitals (eg, clitoral enlargement, fusion of the labia majora, a urogenital sinus rather than distinct urethral and vaginal openings); later in life they may have hirsutism, oligomenorrhea, and acne.
In males, androgen excess may not be apparent or may manifest in childhood with increased growth velocity and early signs of puberty.
In both sexes, salt wasting causes hyponatremia and hyperkalemia.
Diagnose by steroid hormone levels and sometimes adrenocorticotropic hormone (ACTH) stimulation and/or genotyping.
Treat with replacement of glucocorticoids and sometimes mineralocorticoids.
Drug Information for the Topic





