





Sickle cell disease is an inherited genetic abnormality of hemoglobin (the oxygen-carrying protein found in red blood cells) characterized by sickle (crescent)-shaped red blood cells and chronic anemia caused by excessive destruction of the abnormal red blood cells.
People have anemia and sometimes jaundice.
Pain in the long bones, abdomen, and chest can indicate sickle cell crisis.
Worsening anemia, fever, shortness of breath, and low oxygen levels can indicate acute chest syndrome, a major complication of sickle cell disease.
A special blood test called electrophoresis can be used to determine whether people have sickle cell disease.
Avoiding activities that may cause crises and treating infections and other disorders quickly can help prevent crises.
Sickle cell disease is an inherited genetic abnormality of hemoglobin in red blood cells that causes them to be misshapen and prone to early destruction by the body. They clog small vessels and block blood flow to tissues, causing pain and organ damage.
Sickle cell disease most frequently affects people with African or Black American ancestry. People who have 1 copy of the gene for sickle cell disease have sickle cell trait. People who have sickle cell trait do not develop sickle cell disease, but they do have increased risks of some complications such as blood in their urine. People who have 2 copies of the gene develop sickle cell disease.
Red Blood Cell Shapes
Normal red blood cells are flexible and disk-shaped, thicker at the edges than in the middle. In several hereditary disorders, red blood cells become spherical (in hereditary spherocytosis), oval (in hereditary elliptocytosis), or sickle-shaped (in sickle cell disease). |

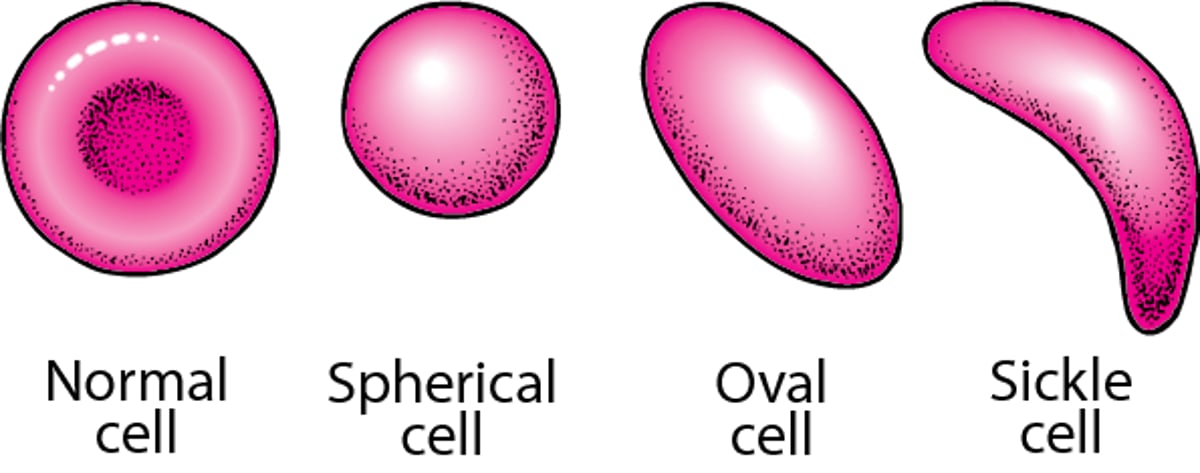
In sickle cell disease, the red blood cells contain an abnormal form of hemoglobin (the protein that carries oxygen). The abnormal form of hemoglobin is called hemoglobin S. When red blood cells contain hemoglobin S, they can become deformed into a sickle shape and less flexible. Not every red blood cell is sickle-shaped. The sickle-shaped cells become more numerous when people have infections or low levels of oxygen in the blood.
The sickle cells are fragile and have shortened survival. Damaged cells are also cleared from the circulation. Because the sickle cells are stiff and stick together, they have difficulty traveling through the smallest blood vessels (capillaries), blocking blood flow and reducing oxygen supply to tissues in areas where capillaries are blocked. The blockage of blood flow can cause pain and, over time, cause damage to the spleen, kidneys, brain, bones, and other organs. Kidney failure and heart failure may occur.


Symptoms of Sickle Cell Disease
People who have sickle cell disease always have some degree of anemia (often causing fatigue, weakness, and paleness) and may have mild jaundice (yellowing of the skin and whites of the eyes). Some people have few other symptoms. Others have severe, recurring symptoms that cause enormous disability and early death.
Sickle cell trait
In people with sickle cell trait, red blood cells are not fragile and do not break easily. Sickle cell trait does not cause painful crises.
People with sickle cell trait are at increased risk of chronic kidney disease and pulmonary embolism. Rarely, they may notice blood in their urine. People with sickle cell trait are also at risk for an extremely rare form of kidney cancer. Individuals with sickle cell trait have normal life expectancy.
Sickle cell crisis
Anything that reduces the amount of oxygen in the blood, cold temperatures, or an illness, may bring on a sickle cell crisis (also called an exacerbation). A sickle cell pain (vaso-occlusive) crisis is an episode of increased symptoms and can consist of a worsening of anemia, pain (often in the chest or long bones of the arms and legs), sometimes shortness of breath, and deterioration of the function of multiple organs. Abdominal pain may be severe, and vomiting may occur and should prompt consideration of splenic or hepatic sequestration. Sometimes, additional complications occur, including:
Aplastic crisis: Production of red blood cells in the bone marrow stops during infection with some viruses
Acute chest syndrome: Caused by blockage of capillaries in the lungs or infection
Acute splenic or hepatic (liver) sequestration (a large accumulation of cells in an organ): Rapid enlargement of the spleen or liver
Acute chest syndrome can occur in people of all ages, but it is most common among children. It is usually characterized by severe pain and difficulty breathing. Acute chest syndrome can be fatal.
In children, acute sequestration of sickled cells in the spleen (sequestration crisis) may occur causing an enlarged spleen and worsening anemia. Acute hepatic (liver) sequestration is less common and can occur at any age.

Complications
Most people who have sickle cell disease develop an enlarged spleen during childhood because sickled cells become trapped in the spleen. By the time the person reaches adolescence, the spleen is often so badly injured that it shrinks and no longer functions. Because the spleen helps fight infection, people with sickle cell disease are more likely to develop pneumococcal pneumonia and other infections. Viral infections, in particular, can decrease red blood cell production, so anemia becomes more severe.
The liver can become progressively larger throughout life (causing upper abdominal fullness), and gallstones often form from the pigment of broken-apart red blood cells.
The heart usually enlarges, and an enlarged heart is less effective in pumping blood to the body, possibly leading to heart failure. Heart murmurs are common.
Children who have sickle cell disease often have a relatively short torso but long arms, legs, fingers, and toes. Changes in the bones and bone marrow may cause bone pain, especially in the hands and feet. Episodes of joint pain may occur, and the hip joint may become so damaged that it eventually needs to be replaced.
Poor circulation to the skin may cause sores on the legs, especially at the ankles. Young males may develop persistent, often painful erections (priapism). Episodes of priapism may permanently damage the penis so that erections can no longer occur. Blocked blood vessels may cause strokes that damage the nervous system. In older adults, lung and kidney function may deteriorate.
Diagnosis of Sickle Cell Disease
Blood tests
Hemoglobin electrophoresis
Prenatal testing
When doctors suspect sickle cell disease, they do blood tests. Sickle-shaped red blood cells and fragments of destroyed red blood cells can be seen in a blood sample examined under a microscope.

Hemoglobin electrophoresis, another blood test, is also done. In electrophoresis, an electrical current is used to separate the different types of hemoglobin and thus detect abnormal hemoglobin.
Further testing may be needed, depending on the specific symptoms the person experiences during the crisis. For example, if the person has difficulty breathing or a fever, a chest x-ray may be done.
Screening for Sickle Cell Disease
Blood tests are done on relatives of people with the disorder because they also may have sickle cell disease or trait. Discovering the trait in people may be important for family planning, to determine their risk of having a child with sickle cell disease.
In the United States, newborns are routinely screened for sickle cell disease or trait with a blood test.
Tests can be done during early pregnancy to screen the fetus and allow prenatal counseling for couples who are at risk of having a child with sickle cell disease. Fetal cells obtained through amniocentesis or chorionic villus sampling are tested for the presence of the sickle cell gene.
Treatment of Sickle Cell Disease
Treatments aimed at preventing crises
Treatment of crises and conditions that cause them
Treatment is aimed at:
Preventing crises
Controlling the anemia
Relieving symptoms
Stem cell transplantation may cure sickle cell disease. Bone marrow or stem cells from a family member or other donor who does not have sickle cell disease may be transplanted in a person with the disease. Such transplantation may be curative.
Gene therapy, a technique in which normal genes are implanted in precursor cells (cells that produce blood cells), has become available. Gene therapy may be curative.
Preventing sickle cell crisis
People who have sickle cell disease should try to avoid activities that reduce the amount of oxygen in their blood and should seek prompt medical attention for even minor illnesses, such as viral infections. Because people are at increased risk of infection, they should receive vaccines for pneumococcal, meningococcal, influenza, and Haemophilus influenzae type b infections. COVID-19 vaccination is also recommended. Children typically take penicillin by mouth until at least age 5 years.
Medications can help control sickle cell disease. For example, hydroxyurea increases the production of a form of hemoglobin found predominantly in fetuses, which decreases the number of red blood cells becoming sickle-shaped. Therefore, it reduces the frequency of sickle cell crises and acute chest syndrome.
Other medications that may help control the symptoms and complications of sickle cell disease are L-glutamine and crizanlizumab.
Controlling anemia
People are given folic acid, a vitamin that helps the body make new red blood cells.
Blood transfusions may be given to correct the anemia.
Treating sickle cell crisis
Sickle cell crisis may require hospitalization. People are given oxygen and fluids by vein (intravenously), as needed, and medications to relieve pain. Blood transfusions and oxygen may be given. Conditions that may have caused the crisis, such as an infection, are treated.
More Information
The following English-language resource may be useful. Please note that The Manual is not responsible for the content of this resource.
Drug Information for the Topic





