



Huntington disease is a hereditary disease that causes random, flowing movements that are involuntary and cannot be suppressed (called chorea). Sometimes a muscle or a group of muscles jerk (called myoclonic jerks). The disorder progresses to more pronounced involuntary movements (sometimes writhing movements, called athetosis), mental deterioration, and death.
In Huntington disease, parts of the brain that help smooth and coordinate movements degenerate.
Movements become jerky and uncoordinated, and mental function, including self-control and memory, deteriorates.
Doctors base the diagnosis on symptoms, family history, imaging of the brain, and genetic testing.
Medications can help relieve the symptoms, but the disorder is progressive, ultimately ending in death.
(See also Overview of Movement Disorders.)
Huntington disease affects about 4 in 100,000 people worldwide. It affects both sexes equally.
The gene for Huntington disease is dominant. That is, having only 1 copy of the abnormal gene, inherited from 1 parent, is sufficient to cause the disease. Therefore, children of a person who has Huntington disease have a 50% chance of developing it.
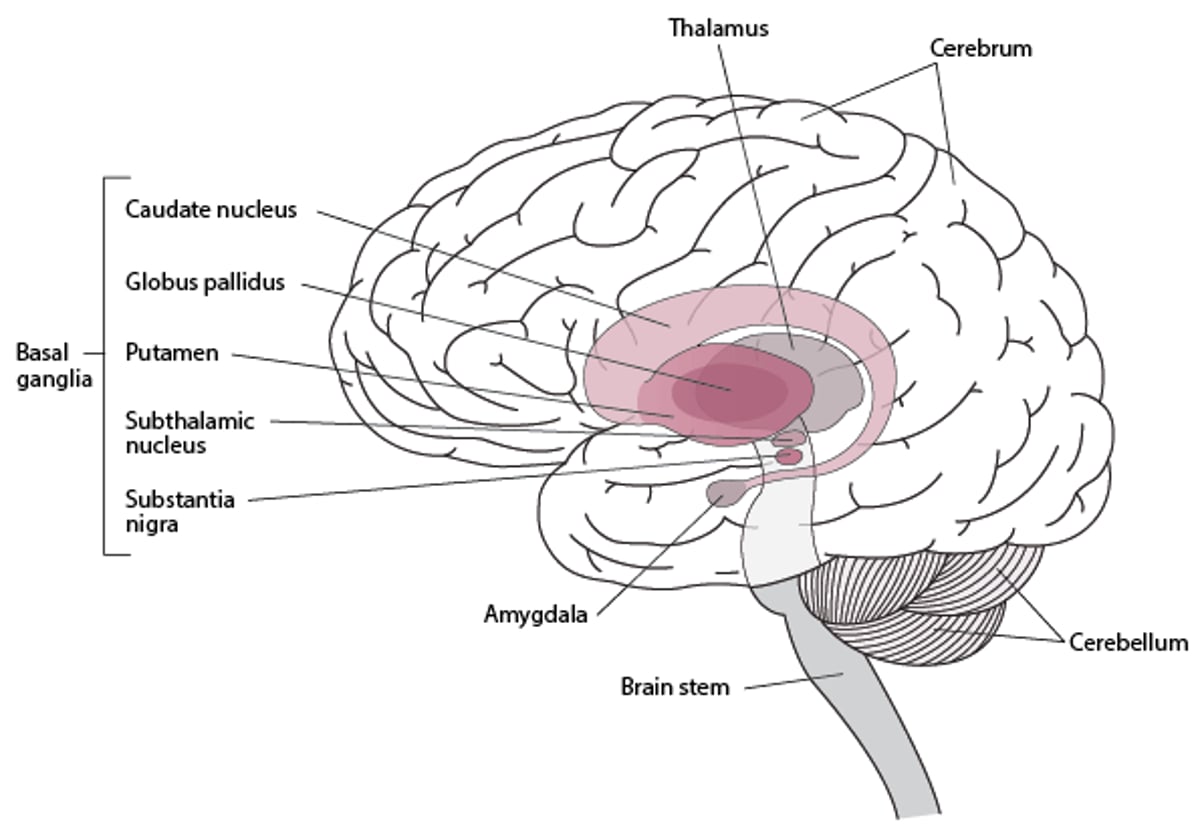
Huntington disease is caused by gradual degeneration of parts of the basal ganglia called the caudate nucleus and putamen. The basal ganglia are collections of nerve cells located at the base of the cerebrum, deep within the brain. They help smooth out and coordinate movements.
Locating the Basal Ganglia
The basal ganglia are collections of nerve cells located deep within the brain. They include the following:
The basal ganglia help initiate and smooth out muscle movements, suppress involuntary movements, and coordinate changes in posture. |

Symptoms of Huntington Disease
Symptoms of Huntington disease usually develop subtly, typically beginning between the ages of 35 and 40 but sometimes before adulthood.
During the early stages of Huntington disease, the face, trunk, and limbs may move involuntarily and rapidly. At first, people can blend these abnormal involuntary movements into purposeful ones so that the abnormal movements are barely noticeable. However, with time, the movements become more obvious.
Muscles may contract briefly and rapidly, causing the arms or another body part to suddenly jerk, sometimes several times in a row.
People may walk in a lilting or exaggeratedly jaunty way, like a puppet. They may grimace, flick the limbs, or repeatedly blink. Movements become uncoordinated and slow. Eventually, the entire body is affected, making walking, sitting still, eating, speaking, swallowing, and dressing extremely difficult.
Mental changes frequently occur before or as the abnormal movements develop. These changes are subtle at first. People may gradually become irritable, excitable, and agitated. They may lose interest in their usual activities. They may be unable to control their impulses, losing their temper, having fits of despondency, or becoming promiscuous.
As Huntington disease progresses, people may behave irresponsibly and often wander aimlessly. Over years, they lose their memory and their ability to think rationally. They may become severely depressed and attempt suicide. They may also become anxious or develop psychiatric conditions such as obsessive-compulsive disorder, bipolar disorder, or schizophreniform disorder.
In advanced disease, dementia is severe, and people are confined to bed. Full-time assistance or nursing home care is needed. Death usually occurs 13 to 15 years after symptoms begin.
Diagnosis of Huntington Disease
A doctor's evaluation, confirmed by genetic testing
Computed tomography or magnetic resonance imaging of the brain
Huntington disease may be difficult to recognize in the early stages because symptoms are subtle. The disease may be suspected based on symptoms and a family history. Doctors should be told about relatives who have had mental problems or have been diagnosed as having a neurologic disorder (such as Parkinson disease) or a psychiatric disorder (such as schizophrenia) because they may have had Huntington disease that was not diagnosed.
Computed tomography (CT) or magnetic resonance imaging (MRI) is done to check for the degeneration of the basal ganglia and other areas of the brain usually affected by the disease and to rule out other disorders.
Genetic testing is done to confirm the diagnosis. Genetic testing and counseling are important for people who have a family history of the disease but no symptoms because people are likely to have children before symptoms appear. For such people, genetic counseling should precede genetic testing. They are referred to centers that have expertise in dealing with the complex ethical and psychological issues involved.
Treatment of Huntington Disease
Antipsychotic and other medications to relieve symptoms
As soon as possible after the diagnosis is made, people with Huntington disease should establish advance directives, indicating what kind of medical care they want at the end of life.
No cure exists for Huntington disease. However, certain medications may help lessen symptoms. Antipsychotic medications (such as chlorpromazine, haloperidol, risperidone, and olanzapine) may help people with depression and control any agitation. Medications that reduce the amount of (such as chlorpromazine, haloperidol, risperidone, and olanzapine) may help people with depression and control any agitation. Medications that reduce the amount ofdopamine (such as tetrabenazine, deutetrabenazine, and the antihypertensive reserpine), can help stop (suppress) the abnormal movements.(such as tetrabenazine, deutetrabenazine, and the antihypertensive reserpine), can help stop (suppress) the abnormal movements.
Antidepressants can also be used to treat depression.
Doctors offer genetic counseling and genetic testing to people who have parents or siblings with Huntington disease. Genetic counseling should be offered before genetic testing because the consequences of Huntington disease are so serious. Counseling is particularly important for women of childbearing age and men considering becoming fathers.
More Information
The following English-language resource may be useful. Please note that The Manual is not responsible for the content of this resource.
Drug Information for the Topic





