



Atherosclerosis is characterized by the development of fatty and/or fibrous intimal plaques (atheromas) in the arterial wall. Initially, the plaques contain lipid materials, but over time they may become fibrosed and calcified. The key pathophysiological processes in the development of atherosclerosis involve low-density lipoprotein (LDL) particles, inflammatory cells, endothelial dysfunction, smooth muscle cell proliferation, and remodeling of extracellular matrix. Common risk factors include age, family history of premature atherosclerotic disease, dyslipidemia, cardiovascular-kidney metabolic factors (eg, diabetes, hypertension, obesity, chronic kidney disease), inflammation, and lifestyle factors (eg, cigarette smoking,a sedentary lifestyle). Symptoms develop when the growth or rupture of the plaque reduces or obstructs blood flow; specific symptoms depend on the affected artery. Diagnosis is primarily clinical and is confirmed by angiography, ultrasound, computed tomography, or other imaging tests. Treatment includes lifestyle changes, addressing established risk factors through the use of lipid-lowering medications, antiplatelet medications, and antiatherogenic medications, as well as catheter-based and surgical interventions.
Atherosclerosis can affect all large and medium-sized arteries, including the coronary, carotid, and cerebral arteries, the aorta and its branches, and the major arteries of the extremities. According to the World Health Organization (WHO), ischemic heart disease (caused by atherosclerosis of the coronary arteries) is the world's leading cause of death, accounting for 13% of all global fatalities between 2000 and 2021 (1). Since 2000, ischemic heart disease has had the largest surge in fatalities, increasing by 2.7 million to reach a total of 9.0 million deaths in 2021. In 2022, in the United States, approximately 942,000 people died of cardiovascular disease; heart disease and stroke caused more deaths than the combined total of all cancer types and chronic lower respiratory diseases (2). Although ischemic heart disease continues to be a leading cause of mortality, advancements in cardiovascular care and prevention have resulted in significant declines in age-standardized cardiovascular mortality since 1950 (3, 4). However, a worrisome rapid increase in the prevalence of atherosclerosis has been noted in low and middle income countries, in part due to an increase in hypertension, chronic disease, and overall caloric intake (5). Despite advances in care, atherosclerosis remains the leading cause of death worldwide.

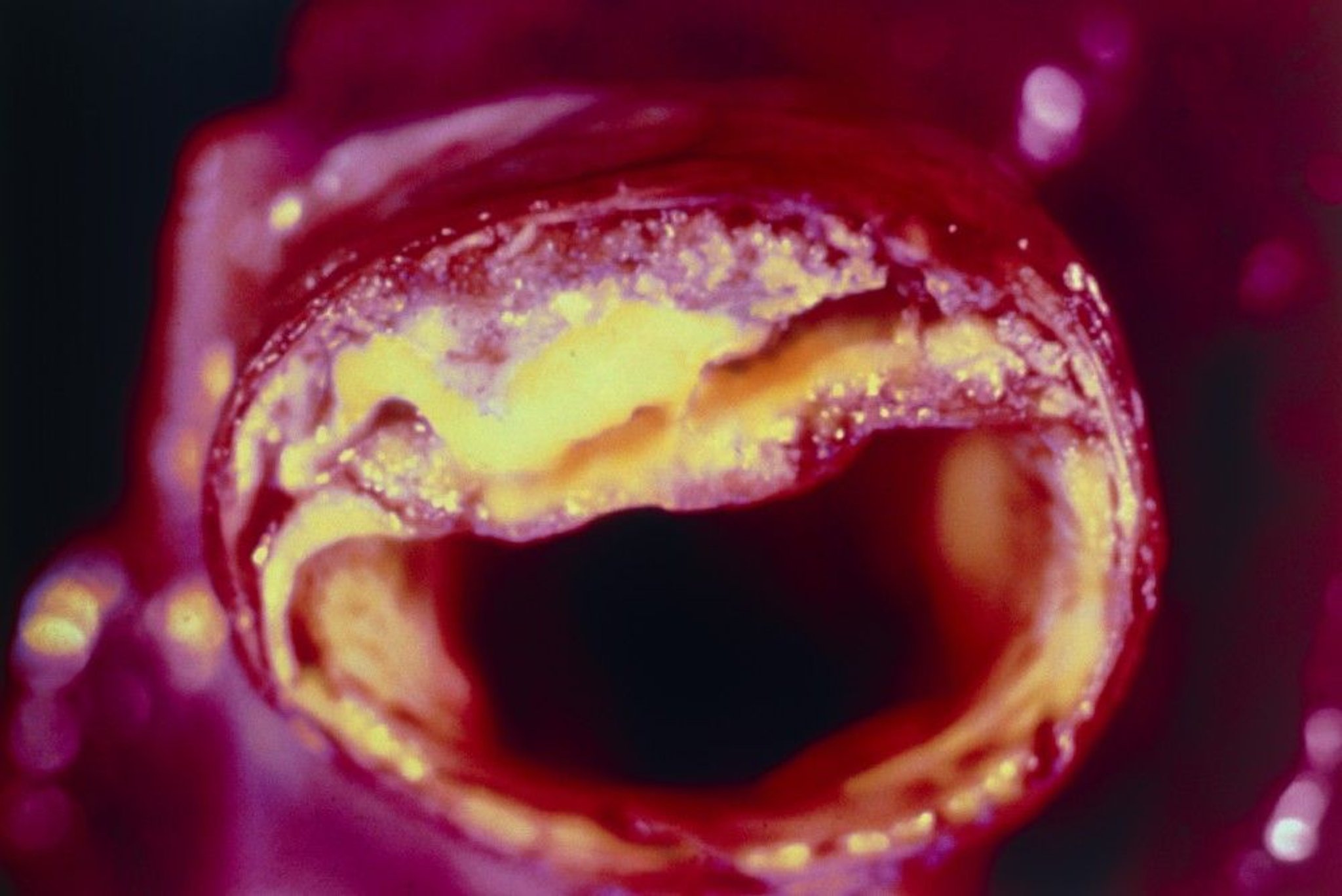
Cross-section of an artery with atheroma visible in the upper section.
BSIP VEM/SCIENCE PHOTO LIBRARY
General references
1. World Health Organization: The Global Health Observatory: Global health estimates: Leading causes of death. Cause-specific mortality, 2000–2021. Accessed August 4, 2025.
2. Martin SS, Aday AW, Allen NB, et al. 2025 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation. 2025;151(8):e41-e660. doi:10.1161/CIR.0000000000001303
3. Fox CS, Evans JC, Larson MG, Kannel WB, Levy D. Temporal trends in coronary heart disease mortality and sudden cardiac death from 1950 to 1999: the Framingham Heart Study. Circulation. 2004;110(5):522-527. doi:10.1161/01.CIR.0000136993.34344.41
4. Mensah GA, Wei GS, Sorlie PD, et al. Decline in Cardiovascular Mortality: Possible Causes and Implications. Circ Res. 2017;120(2):366-380. doi:10.1161/CIRCRESAHA.116.309115
5. Li Y, Cao GY, Jing WZ, Liu J, Liu M. Global trends and regional differences in incidence and mortality of cardiovascular disease, 1990-2019: findings from 2019 global burden of disease study. Eur J Prev Cardiol. 2023;30(3):276-286. doi:10.1093/eurjpc/zwac285
Pathophysiology of Atherosclerosis
The atherosclerotic plaque is the hallmark feature of atherosclerosis. It has a complex and dynamic structure with the following major components:
Lipid core: Consists mainly of cholesterol, cholesterol esters, and other lipids
Fibrous cap: A layer of smooth muscle cells and collagen that covers the lipid core
Inflammatory cells and smooth muscle cells: Macrophages, T cells, and other immune cells that infiltrate the plaque
Cell debris: Dead or necrotic cells
Calcium deposits: Contribute to the rigidity of the plaque
Extracellular matrix: Includes elastin, collagen, and other macromolecules, providing structural support
Atherosclerotic plaque formation
All stages of atherosclerosis, from initiation and growth to plaque complication (eg, myocardial infarction, stroke), are considered a cytokine-mediated inflammatory response to injury (such as shear stress from hypertension, or oxidative injury from tobacco smoke) (1).
During the initial stages of atherosclerotic plaque formation, apolipoprotein B-containing lipoproteins, particularly low density lipoprotein (LDL) particles, accumulate in the intima, the innermost layer of the arterial wall. Within the intimal layer, LDL particles are shielded from the protective antioxidant mechanisms of the plasma, and, as a result, they undergo oxidation and other modifications mediated by oxidative, lipolytic, and proteolytic enzymes, as well as reactive oxygen species. These chemical modifications lead to a proinflammatory and immunogenic switch in phenotype.
This event triggers the migration of proinflammatory, classic monocytes into the intimal layer, facilitated by adhesion molecules (eg, vascular cell adhesion molecule-1 [VCAM-1]) expressed on the surface of activated endothelial cells. The local abundance of chemoattractant cytokines facilitates the movement of these bound monocytes into the arterial wall. Inside the intima, monocytes transform into macrophages, expressing scavenger receptors that allow them to ingest lipoprotein particles and become foam cells, a hallmark of the early atherosclerotic plaque. The migration of T cells also plays a significant role in establishing the inflammatory environment of the developing atherosclerotic plaque (2). In response to signals from the accumulating leukocytes, smooth muscle cells originating from the tunica media also migrate into the intimal layer, augmenting the population of resident intimal smooth muscle cells.
As atherosclerotic plaques evolve, both resident and recruited smooth muscle cells produce extracellular matrix molecules, including collagen, elastin, proteoglycans, and glycosaminoglycans, resulting in intimal thickening (3). Within the maturing lesion, smooth muscle cells and macrophages proliferate. These smooth muscle cells and macrophages may also undergo necrosis or apoptosis (programmed cell death), leading to the accumulation of cell debris and forming a necrotic, lipid-rich core.
Certain chemokines, such as interferon gamma released by T cells, can inhibit the ability of smooth muscle cells to produce interstitial collagen, thereby weakening their capacity to repair and sustain the fibrous cap that overlays the necrotic core. Additionally, activated macrophages increase the production of matrix metalloproteinases, a family of enzymes that can degrade the interstitial collagen vital for the fibrous cap's strength. This degradation results in the thinning and weakening of the fibrous cap, making the plaque more prone to rupture.
Stable and Vulnerable Atherosclerotic Plaques
Top: A normal artery (left) compared with an artery that is narrowed by atherosclerotic plaque (right). Bottom: A vulnerable plaque (left) with a thin fibrous cap and a large lipid core compared to a stable plaque (right) with a thicker fibrous cap and smaller lipid core. |


Plaque stability and rupture
Atherosclerotic plaques may be stable or unstable.
Stable plaques either regress, remain static, or grow slowly over several decades, potentially causing vascular stenosis or occlusion. During the progression of the atherosclerotic lesion, the arterial wall expands outwardly to preserve the vascular lumen (positive remodeling, also known as the Glagov phenomenon) (4). This outward remodeling can mask the severity of atherosclerosis, allowing significant plaque buildup without narrowing that is detectable with angiography.
Unstable, or "vulnerable," plaques are prone to spontaneous erosion or rupture, leading to acute thrombosis, occlusion, and infarction, often long before they cause hemodynamically significant stenosis. These plaques typically feature a large lipid core covered by a thin (< 60 microns) fibrous cap. Most clinical events result from unstable plaques that do not appear hemodynamically significant when examined with angiography. Therefore, plaque stabilization is a critical strategy to reduce morbidity and mortality. (See figure .)
Plaque complications include plaque rupture and erosion.
Plaque rupture is the most common cause of acute coronary thrombosis leading to acute myocardial infarctions (5, 6), particularly fatal ones.
The strength of the fibrous cap and its resistance to rupture depend on the balance between collagen deposition and degradation. Plaque rupture involves secretion of metalloproteinases, cathepsins, and collagenases by activated macrophages within the plaque. These enzymes digest the fibrous cap, particularly at its edges, causing the cap to thin and ultimately rupture. T cells in the plaque contribute by secreting cytokines that inhibit smooth muscle cells from synthesizing and depositing collagen, which normally reinforces the plaque.
Once the plaque ruptures, the thrombogenic contents of the plaque are exposed to circulating blood, triggering thrombosis. Tissue factor produced by macrophages and smooth muscle cells significantly contributes to this process by promoting thrombin generation in vivo, leading to thrombin-mediated fibrin formation from fibrinogen and activation of platelet aggregation. One of 4 outcomes may occur:
The resultant thrombus may organize and be incorporated into the plaque, altering its shape and causing its rapid growth.
The thrombus may rapidly occlude the vascular lumen, precipitating an acute ischemic event.
The plaque may fill with blood, balloon out, and immediately occlude the artery.
Plaque contents or the thrombus may embolize, occluding vessels downstream.
Plaque stability depends on multiple factors, including plaque composition (relative proportions of lipids, inflammatory cells, smooth muscle cells, connective tissue, and thrombus), wall stress (cap fatigue), size and location of the core, and the configuration of the plaque in relation to blood flow. Intraplaque hemorrhage, by contributing to rapid growth and lipid deposition, may play an important role in transforming stable plaques into unstable plaques.
In general, unstable coronary artery plaques have a high macrophage content, a thick lipid core, and a thin fibrous cap; they tend to rupture unpredictably, and often narrow the vessel lumen by < 50% (7, 8). Unstable carotid artery plaques have a similar composition but typically cause problems through severe stenosis and occlusion or by deposition of platelet thrombi, which embolize rather than rupture. Low-risk plaques have a thicker cap and contain fewer lipids; they often narrow the vessel lumen by > 50% and may produce predictable exercise-induced stable angina.
Clinical consequences of plaque rupture in coronary arteries depend not only on the anatomical location of the plaque but also on the relative balance of procoagulant and anticoagulant activity in the blood, as well as the vulnerability of the myocardium to arrhythmias.
Plaque erosion is the second most common underlying mechanism for acute coronary syndromes, accounting for about one-third of the cases (and about two-thirds of non-ST-segment elevation myocardial infarctions) (9, 10). Unlike plaque rupture, plaque erosion happens without breaking the fibrous cap of the atherosclerotic plaque. The primary underlying mechanism is the loss of the endothelial cells over an intact fibrous cap, exposing the underlying plaque material to the circulating blood and triggering thrombosis. Erosion has been associated with the activation of innate immune cells, especially through pattern-recognition receptors such as Toll-like receptor 2, and the activation of polymorphonuclear leukocytes leading to the destruction of the endothelial lining (11). Lesions complicated by plaque erosion tend to have a rich extracellular matrix with less lipid content and do not exhibit a vulnerable thin fibrous cap.
Risk Factors for Atherosclerosis
There are numerous risk factors for atherosclerosis (see table ) (1), both modifiable and non-modifiable.
Risk Factors for Atherosclerosis
Status | Risk Factor |
|---|---|
Nonmodifiable | Age Family history of premature atherosclerotic cardiovascular disease and genetic predisposition* Male sex† Ethnicity (eg,South Asian ancestry) |
Modifiable | Dyslipidemia (lipid abnormalities):
Cardiovascular-kidney-metabolic factors:
Inflammation:
Infection:
Lifestyle:
Other:
|
* Atherosclerotic cardiovascular disease is considered premature when it occurs in a male first-degree relative before age 55 years and in a female first-degree relative before age 65 years. Atherosclerotic cardiovascular disease includes coronary artery disease (myocardial infarction, angina or coronary artery stenosis), cerebrovascular disease (transient ischemic attack, ischemic stroke, carotid stenosis), peripheral artery disease, and aortic atherosclerotic disease. | |
† Atherosclerosis is increasingly prevalent in young women and women members of minority groups and populations in developing countries. | |
Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;74(10):e177-e232. doi:10.1016/j.jacc.2019.03.010; Hamad R, Penko J, Kazi DS, et al. Association of Low Socioeconomic Status With Premature Coronary Heart Disease in US Adults. JAMA Cardiol. 2020;5(8):899-908. doi:10.1001/jamacardio.2020.1458; Izquierdo-Condoy JS, Arias-Intriago M, Becerra Cardona DA, García-Cañarte S, Vinueza-Moreano P. Anticancer Chemotherapy-Induced Atherosclerotic Cardiovascular Disease: A Comprehensive Review. Life (Basel). 2025;15(2):245. doi:10.3390/life15020245; Levine GN, Cohen BE, Commodore-Mensah Y, et al. Psychological Health, Well-Being, and the Mind-Heart-Body Connection: A Scientific Statement From the American Heart Association. Circulation. 2021;143(10):e763-e783. doi:10.1161/CIR.0000000000000947; Libby P, Buring JE, Badimon L, et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5(1):56. doi:10.1038/s41572-019-0106-z; Pandey AK, Blaha MJ, Sharma K, et al. Family history of coronary heart disease and the incidence and progression of coronary artery calcification: Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis. 2014;232(2):369-376. doi:10.1016/j.atherosclerosis.2013.11.042; Szwed P, Gąsecka A, Zawadka M, et al. Infections as Novel Risk Factors of Atherosclerotic Cardiovascular Diseases: Pathophysiological Links and Therapeutic Implications. J Clin Med. 2021;10(12):2539. doi:10.3390/jcm10122539. | |
CRP = C-reactive protein, HDL = high-density lipoprotein cholesterol, LDL = low-density lipoprotein cholesterol. | |
Nonmodifiable risk factors
Age alone is a significant risk factor for the development of atherosclerosis. There is a dramatic increase observed in the prevalence of peripheral artery disease, coronary artery disease and abdominal aortic aneurysms with advanced age (2).
Male sex is a well-established risk factor for earlier and more severe atherosclerotic cardiovascular disease (3). While younger women generally have a lower risk of atherosclerosis, by the time they reach their seventies, the incidence of myocardial infarction in women exceeds that of men, indicating an interaction between sex and age (2).
Patients with a family history of premature cardiovascular disease also exhibit increased risk for development of atherosclerosis (4). The list of genetic disorders documented in the pathogenesis of atherosclerosis is growing and includes both monogenetic conditions such as familial hypercholesterolemia associated with LDL receptor gene mutations, and polygenetic heredity.
There are significant ethnic differences in atherosclerosis risk. For example, people of South Asian ancestry show a substantially higher risk of atherosclerotic cardiovascular disease than people of European ancestry (5).
Modifiable risk factors
Dyslipidemia
In the presence of dyslipidemia, apolipoprotein B-containing particles (mainly LDL) accumulate in the intimal layer of the arteries where they undergo oxidation and switch to a pro-inflammatory phenotype. This event leads to the migration and activation of inflammatory cells, primarily monocytes and T cells (6). As a result, LDL plays a central role in the development of atherosclerosis. A growing body of literature suggests that the risk for developing atherosclerotic cardiovascular disease is proportional to the cumulative exposure to LDL cholesterol, often referred to as cholesterol years (7). Additionally, the concentration of small, dense, lipid-depleted LDL particles is a significant risk factor for atherosclerotic cardiovascular disease (ASCVD) (8).
The main determinant for risk of ASCVD, however, is the concentration of atherogenic lipoprotein particles, which is best reflected by the apolipoprotein B (ApoB) concentration, or by the non-high density lipoprotein (HDL)-cholesterol concentration if a specific ApoB test is unavailable (9). For risk estimation, ApoB generally provides a more accurate and consistent prediction, especially in cases of discordance between ApoB and LDL cholesterol (10). ApoB-100 is capable of binding the LDL receptor and is responsible for cholesterol transport. It is also responsible for transporting oxidized phospholipids and possesses proinflammatory properties.
HDL has been traditionally viewed as a protective factor against atherosclerosis by facilitating reverse cholesterol transport. Studies indicate a U-shaped association between cardiovascular risk and HDL levels, suggesting that individuals with both the lowest (< 40 mg/dL [< 1.0 mmol/L ] in men and < 50 mg/dL [< 1.3 mmol/L] in women) and highest (approximately 80 to 100 mg/dL [2.07 to 2.59 mmol/L]) HDL cholesterol levels face an increased risk of cardiovascular mortality (11, 12) (see table ).
Lipoprotein (a) [Lp(a)] is a pro-atherogenic lipoprotein that consists of an LDL-like core associated with an additional protein, apolipoprotein(a), which is covalently linked with the ApoB-100 molecule. Elevated Lp(a) levels confer an independent risk for atherosclerotic cardiovascular disease (13). Lp(a) levels are largely genetically determined and remain fairly stable throughout life.
Triglyceride-rich lipoproteins also play a significant role in the development of atherosclerosis. Hypertriglyceridemia is an independent risk factor for atherosclerotic events, even in patients whose LDL levels are adequately controlled by statin therapy (residual triglyceride risk) (14).
Cardiovascular-kidney-metabolic factors
Cardiovascular-kidney-metabolic (CKM) syndrome is an interconnected health disorder involving obesity, diabetes, chronic kidney disease, and cardiovascular disease (15). CKM syndrome includes individuals at risk for atherosclerosis as well as those with existing atherosclerotic cardiovascular disease, with the following CKM stages (15):
Stage 0: No CKM risk factors
Stage 1: Excess or dysfunctional adiposity
Stage 2: Metabolic risk factors such as hypertriglyceridemia, hypertension, diabetes, metabolic syndrome, or moderate- to high-risk chronic kidney disease
Stage 3: Subclinical cardiovascular disease in CKM syndrome or risk equivalents, such as high predicted cardiovascular risk or very high-risk chronic kidney disease (CKD)
Stage 4: Clinical cardiovascular disease in CKM syndrome
Diabetes leads to formation of advanced glycation end products, which increase the production of proinflammatory cytokines from endothelial cells (16). The oxidative stress and reactive oxygen radicals generated in diabetes directly injure the endothelium and promote atherogenesis.
Hypertension is a well known risk factor for atherosclerosis. However, the underlying mechanisms are not well established. Among other mechanisms, endothelial cell activation, oxidative stress, and the contribution to smooth muscle cell proliferation have been implicated (17). ASCVD risk increases above a threshold blood pressure as low as 115 mmHg (systolic) and 75 mmHg (diastolic), levels that do not fall within the hypertensive range (18). Risk then continues to increase roughly linearly as blood pressure increases (19, 20).
Chronic kidney disease promotes the development of atherosclerosis through several pathways, including worsening hypertension, insulin resistance, and increased levels of lipoprotein(a), homocysteine, fibrinogen, and C-reactive protein (21).
Inflammation
Elevated C-reactive protein (CRP) level (high sensitivity CRP ≥ 2 mg/L) has been associated with an increased risk of cardiovascular events, even in the presence of a normal lipid profile (or one well-controlled on medication even if not completely normal), representing a "residual inflammatory risk" (22). CRP is produced by the liver as a primary acute phase reactant and is involved in platelet activation and the regulation of the innate immune mechanisms.
Autoimmune diseases are associated with an increased risk of developing atherosclerosis independent of traditional risk factors (age, sex, total cholesterol, HDL cholesterol, blood pressure, diabetes, and smoking) (23). The strongest associations have been described in conditions such as rheumatoid arthritis (24), systemic lupus erythematosus, Addison disease, and type 1 diabetes.
Clonal hematopoiesis of indeterminate potential (CHIP), characterized by the presence of an expanded somatic blood-cell clone in individuals without other hematologic abnormalities, is associated with nearly twice the risk of coronary artery disease and early-onset myocardial infarction (25).
Infection may also play a role in atherogenesis. Individuals with human immunodeficiency virus (HIV) infection are at increased risk for developing myocardial infarction and other atherosclerotic complications due to a higher prevalence of traditional risk factors, immune cell activation, direct viral effects on endothelial cells, altered lipoprotein metabolism, and antiretroviral therapy associated dyslipidemia, and/or insulin resistance (26). Infections such as Chlamydia pneumoniae, cytomegalovirus, Helicobacter pylori, severe acute respiratory syndrome coronavirus-2 (COVID-19), influenza, respiratory syncytial virus, those associated with periodontal disease, and others may cause endothelial dysfunction through direct infection, exposure to endotoxins, or stimulation of systemic or subendothelial inflammation (27).
Lifestyle
Tobacco smoke contains nicotine and other chemicals that are toxic to vascular endothelium. Smoking, including passive smoking, increases platelet reactivity (potentially promoting platelet thrombosis) and raises plasma fibrinogen levels (28, 29). Smoking increases LDL and decreases HDL levels, promotes lipid peroxidation, induces vasoconstriction, and stimulates smooth muscle cell proliferation.
Sedentary lifestyle, diet, alcohol consumption, chronic stress and hostility, and other psychosocial factors are also lifestyle-related risk factors for atherosclerotic disease.
Other risk factors
Prothrombotic states (see Overview of Thrombotic Disorders) increase the likelihood of atherothrombosis.
Accelerated coronary atherosclerosis is also observed after thoracic radiation therapy (30). Atherosclerosis is likely the result of radiation-induced endothelial injury associated with reactive oxygen species production and intimal proliferation. In addition, certain chemotherapeutic agents such as anthracyclines, taxanes, tyrosine kinase inhibitors, and immune checkpoint inhibitors have been implicated in the induction of oxidative stress, endothelial dysfunction, systemic inflammation, or disrupted lipid metabolism, potentially contributing to the development or worsening of atherosclerosis (31).
Premature menopause has been associated with a significantly increased risk for the development of cardiovascular disease (32). Data suggest that young women with adverse pregnancy outcomes, including preeclampsia, have a higher rate of coronary atherosclerosis compared to women without a documented history of adverse pregnancy outcomes (33, 34).
Documented vascular disease
The presence of atherosclerotic disease in one vascular territory increases the likelihood of disease in other vascular territories. Patients with noncoronary atherosclerotic vascular disease have cardiac event rates comparable to those of patients with known coronary artery disease. As a result, they are considered to have a coronary artery disease risk equivalent and should be treated as aggressively as patients with coronary artery disease (35).
Assessing Overall Risk
Overall ASCVD risk is determined by a patient's individual risk factors. See Diagnosis - Asymptomatic Patients (Screening) for information on determining overall ASCVD risk in individual patients using a risk estimation calculator.
Symptoms and Signs of Atherosclerosis
Atherosclerosis is initially asymptomatic, often for decades. Symptoms and signs develop when lesions impede blood flow (see table ). The symptoms depend on the vascular bed affected and the acuity and severity of blood flow impairment.
Unstable angina, myocardial infarction, ischemic stroke, or resting pain in the limbs may develop when unstable plaques rupture and acutely occlude a major artery, with superimposed thrombosis or embolism. Plaque rupture may also cause sudden death without preceding angina.
Clinical Manifestations of Atherosclerosis
Vascular Bed | Acute Manifestations | Chronic Manifestations |
|---|---|---|
Carotid or cerebral arteries | Transient ischemic attack (TIA) or stroke: Sudden neurologic symptoms (eg, numbness, weakness especially when focal, confusion, aphasia) | Transient ischemic attacks and vascular dementia: Memory loss; confusion; trouble with reasoning, planning, judgment, language, and other thinking abilities |
Coronary arteries | Acute coronary syndrome (unstable angina and myocardial infarction): Angina, dyspnea, nausea, vomiting, sweating, arrhythmia, hypotension, shock, sudden cardiac death | Stable coronary artery disease: Angina, dyspnea, fatigue |
Thoracic aorta | Dissection or thoracic aortic aneurysm rupture: Sudden severe chest or upper back pain, dyspnea, syncope, hypotension, shock | Thoracic aortic aneurysm: Often asymptomatic |
Abdominal aorta | Abdominal aortic aneurysm rupture: Sudden severe abdominal or back pain, dizziness, hypotension, shock | Abdominal aortic aneurysm: Throbbing or deep back pain, pulsing sensation in the abdomen, intermittent claudication |
Mesenteric arteries | Acute mesenteric ischemia: Sudden severe abdominal pain, nausea, vomiting, diarrhea, blood in the stool | Chronic mesenteric ischemia: Postprandial pain, weight loss |
Renal arteries | Renal artery thrombosis: Sudden severe flank pain, hematuria, nausea, vomiting | Renovascular hypertension and chronic kidney disease: Difficult to control hypertension, fluid retention, fatigue |
Lower extremity peripheral arteries | Acute limb ischemia: Severe pain, coldness, numbness, and pallor in the affected limb | Peripheral artery disease: Claudication, rest pain, non-healing ulcers |
Diagnosis of Atherosclerosis
Approach to diagnosis of atherosclerosis depends on the presence or absence of symptoms.
Symptomatic patients
Patients with signs and symptoms of ischemia (see table ) are evaluated for the extent and location of atherosclerotic disease burden and vascular occlusion using various invasive and noninvasive tests, depending on the organ involved (see elsewhere in The Manual).
Noninvasive techniques that can assess atherosclerosis severity and plaque characteristics include:
Ankle-brachial index (ABI): The ABI is a simple, noninvasive test that compares the blood pressure in the ankle with the blood pressure in the arm. It is used for the evaluation of peripheral artery disease.
Vascular ultrasound: Doppler ultrasound is used to visualize atherosclerotic plaques and assess the severity of stenosis. Vascular ultrasound can be used to assess the carotid arteries, renal arteries, abdominal aorta, and peripheral arteries. It offers high temporal resolution without the use of ionizing radiation.
Noncontrast CT, including coronary artery calcium scoring CT: CT is used to evaluate the presence and extent of calcified plaques. It can be used as a dedicated examination (eg, coronary calcium scoring CT with standard ECG-gated image acquisition) or for opportunistic evaluation using CT images acquired for other indications.
CT angiography (CTA): CTA uses iodinated contrast-enhanced CT to evaluate atherosclerotic plaque morphology and detect stenosis severity with high spatial resolution. This technique involves ionizing radiation and can be applied to any vascular bed, making it ideal for small, mobile vascular beds like the coronary arteries.
CT fractional flow reserve (FFR): This technique combines CTA with computational fluid dynamics to assess the physiologic significance of coronary artery stenoses by estimating the pressure drop across a coronary lesion.
Magnetic resonance angiography (MRA): MRA may be done without contrast or with gadolinium-based contrast. It is used to evaluate vascular size and/or stenosis severity without exposing the patient to ionizing radiation. However, this technique has suboptimal spatial resolution for visualizing small, mobile vascular beds.
Positron emission tomography (PET) molecular imaging: This technique uses molecular imaging tracers to detect metabolic activity of atherosclerotic plaques (F18-fluorodeoxyglucose [FDG]) or microcalcifications (F18-sodium fluoride).
Stress testing: Stress testing, which assesses the functional significance of coronary stenosis, includes exercise treadmill test, stress echocardiography, or exercise/pharmacological myocardial perfusion imaging (including single photon emission tomography [SPECT], PET, or magnetic resonance imaging [MRI]). PET and MRI myocardial perfusion imaging can quantify myocardial blood flow, which also aids in the diagnosis of coronary microvascular disease.


This contrast MRA shows severe renal artery stenosis (arrow).
Image courtesy of Attila Feher, MD, PhD.


This FDG-PET scan shows an avid atherosclerotic plaque of the infrarenal aorta (arrow).
Image courtesy of Attila Feher, MD, PhD.
Invasive catheter-based tests that can assess atherosclerosis severity and plaque characteristics include:
Invasive angiography: A catheter is inserted at the origin of the artery being investigated and iodinated contrast is injected. This technique allows the severity of stenosis to be estimated. In addition, therapeutic interventions, including thrombectomy and angioplasty, can be conducted.
Intravascular ultrasound (IVUS): An intravascular ultrasound transducer is used to assess the composition and extent of atherosclerotic plaques and to guide intravascular procedures.
Optical coherence tomography (OCT): This optical imaging-based method can be used to assess the composition and extent of atherosclerotic plaques and guide intravascular procedures.
Fractional flow reserve (FFR): A pressure wire is used to evaluate the hemodynamic significance of a stenosis. FFR is based on the ratio of pressure distal and proximal to a stenosis in the presence of maximal blood flow (hyperemia, typically induced by adenosine). In coronary arteries, an FFR ≤ 0.80 indicates a hemodynamically significant stenosis.
Coronary flow reserve (CFR):-This technique uses a Doppler or a pressure-temperature sensor-tipped guidewire to assess the functional significance of coronary artery disease and microvascular disease by measuring the ratio of maximal flow in a coronary artery to the flow under resting conditions.
Endothelial function testing: A vasoactive agent (eg, acetylcholine) is injected into the coronary arteries to provoke vasospasm and assess the endothelial response.


A (left): Coronary CT angiogram shows extensive mixed plaques, both calcified (green arrow) and predominantly noncalcified (yellow arrows)..
B (center): Invasive coronary angiogram confirms moderate left anterior descending luminal narrowing (arrow), which is hemodynamically significant by fractional flow reserve (FFR) assessment (
D (bottom right): Intravascular ultrasound (IVUS)–guided revascularization. The extent of luminal narrowing is visible in cross-section..
Images courtesy of Attila Feher, MD, PhD.
Patients with symptoms also should be evaluated for atherosclerosis risk factors through the following assessments (1, 2):
History and physical examination, including body mass index and waist circumference
Blood pressure measurement (to assess hypertension)
Fasting lipid profile with lipoprotein(a), apolipoprotein B, and high-sensitivity CRP assessment if indicated
Fasting glucose and glycosylated hemoglobin (HbA1C) levels
Renal function tests (to check for chronic kidney disease)
Smoking status, including passive smoking exposure
Assessment for any history of radiation therapy to the thoracic region
Evaluation of menopausal status and adverse pregnancy outcomes (eg, preeclampsia) in females
Genetic testing for familial hypercholesterolemia (if family history suggests)
Screening for chronic infections that contribute to atherosclerosis, if clinically appropriate
Asymptomatic Patients (Screening)
In children without significant ASCVD risk factors, recommended obesity screening begins at age 2 to 6 years, blood pressure screening at age 3 years, and lipid screening at age 9 to11 years (3, 4, 5, 6).
For adults age 20 to 39 years, the evaluation of traditional ASCVD risk factors (age, sex, total cholesterol, HDL cholesterol, blood pressure, diabetes, and smoking) is recommended. For adults age 40 to 75 years, the pooled cohort equation risk calculator is also recommended to evaluate 10-year ASCVD risk (7).

Other risk estimation calculators are available:
American Heart Association Predicting Risk of Cardiovascular Disease EVENTs (PREVENT) calculator incorporates chronic kidney disease as a significant risk factor (see PREVENT) (8), but this calculator has not been endorsed by any major cardiovascular guidelines.
European Society of Cardiology (ESC) 2021 cardiovascular disease prevention guidelines recommend using the Systemic Coronary Risk Estimation 2 (SCORE2) for individuals aged 40 to 69 and SCORE2-OP (Older Persons) for risk estimation for those over the age of 70 (9, 10).
Calcium scoring CT uses ECG-gated noncontrast CT to quantify calcified atherosclerotic plaque burden and can be used for risk stratification and reclassification, to help make decisions about withholding or recommending statin therapy. For example, in patients with intermediate risk (estimated 10-year ASCVD risk ≥ 7.5% but < 20%) or in selected patients with borderline risk (estimated 10-year ASCVD risk ≥ 5% but < 7.5%), if the calcium score is zero, it is reasonable to withhold statin therapy and reassess in 5 to 10 years as long as the patient does not smoke, does not have diabetes, and has no family history of premature coronary artery disease; if the calcium score is ≥ 100, it is reasonable to initiate statin therapy. A calcium score of zero has an excellent negative prognostic value, with an estimated 1% risk for major adverse cardiovascular events over 10 years (11, 12).


This calcium scoring CT scan shows calcified atherosclerosis in the following structures:
Red arrow: Left anterior descending coronary artery
Blue arrow: Left main coronary artery
Orange arrow: Ascending aorta near the left main ostium
Yellow arrow: Descending aorta
Image courtesy of Attila Feher, MD, PhD.
Prevention and Treatment of Atherosclerosis
Lifestyle changes:
Smoking cessation
Diet
Physical activity
Mental healthcare
Pharmacotherapy for established risk factors:
Lipid-lowering therapies
Antidiabetic or weight loss medications
Antihypertensives
Anti-inflammatory therapy
Antiplatelet medications
Anticoagulation
Thrombolytics
Catheter-based interventions:
Balloon angioplasty
Stenting
Atherectomy
Intravascular lithotripsy
Brachytherapy
Thrombectomy
Surgical intervention:
Bypass surgery
Endarterectomy
Aneurysm repair
Treatment involves aggressive modification of risk factors to slow progression and induce regression of existing plaques. Given the well-established link between dyslipidemia and atherosclerosis, lowering LDL is the cornerstone of the recommended therapy for established atherosclerotic disease (ASCVD).
Lifestyle changes include diet modification, smoking cessation, regular participation in physical activity, stress reduction strategies, and limitation of alcohol use. Medications to treat dyslipidemia and other underlying risk factors such as hypertension, and diabetes are often required. These lifestyle changes and medications directly or indirectly improve endothelial function and reduce inflammation, and thus serve a role both in prevention and treatment of clinical disease.
Lifestyle changes
Smoking cessation
Smoking is one of the most important preventable causes of atherosclerotic cardiovascular disease. In current smokers, a combination of behavioral intervention and pharmacotherapy is recommended to help with tobacco abstinence (1, 2). Pharmacotherapy options include nicotine replacement therapy, varenicline, or bupropion (see also Smoking Cessation).
Dietary changes that reduce cardiovascular risk
Key dietary recommendations for the prevention and treatment of atherosclerosis include (1, 2):
Prioritize the consumption of fruits, vegetables, legumes, nuts, whole grains, and fish
Reduce the intake of saturated fat and trans fat and replace them by polyunsaturated and monounsaturated fats
Reduce the intake of dietary cholesterol and sodium
Limit the intake of simple sugars, refined carbohydrates, sweetened beverages, and processed meats
Limit alcohol consumption
In their prevention guidelines, the European Society of Cardiology provides specific dietary recommendations (1):
Fiber intake: 30 to 45 grams per day, preferably from whole grains
Fruit consumption: At least 200 grams per day (≥ 2 to 3 servings)
Vegetable consumption: At least 200 grams per day (≥ 2 to 3 servings)
Nuts: 30 grams of unsalted nuts per day
Red meat consumption: Reduction to less than 350 to 500 grams per week
Fish consumption: 1 to 2 times per week.
Diets that include increased consumption of fruits, vegetables, legumes, nuts, whole grains, and lean protein (preferably fish) have consistently been shown to improve survival compared to standard diets low in these ingredients (3, 4, 5). Studies also show that improved outcomes are associated with a Mediterranean diet supplemented with extra-virgin olive oil or nuts, daily intake of 5 servings of fruits and vegetables, and intake of polyunsaturated fatty acids (found in plant-based oils, seeds, and fatty fish) and monounsaturated fatty acids (found in oils, avocados, and nuts) (6, 7).
Dietary elements that increase cardiovascular risk
Saturated fats (found in animal products and certain oils) and trans fats (prevalent in processed and fried foods) are associated with increased all-cause mortality, supporting recommendations to replace saturated and trans fats with unsaturated fats in the diet (8). Saturated fats should account for less than 10% of total energy intake, and should be replaced by polyunsaturated and monounsaturated fatty acids, as well as carbohydrates from whole grains (1).
Reduction in dietary sodium intake has been associated with lowered blood pressure and a reduced rate of cardiovascular events. Consuming more than 2 grams of sodium per day is linked to increased cardiovascular mortality (9).
Consuming added sugar exceeding 10% of daily calories has been linked to higher cardiovascular mortality (10). Additionally, the use of sweetened beverages and juices has been associated with an increased rate of coronary events and cardiovascular mortality (4).
The frequency and amount (> 14 g/day) of red and processed meat consumption has been associated with a higher risk of cardiovascular mortality (9).
Several large studies suggest that any amount of alcohol consumption is associated with increased atherosclerotic disease risk, challenging prior data suggesting that a low or moderate level of alcohol consumption is associated with lower risk of, or could even be protective against, atherosclerotic disease (11, 12, 13). Current recommendations include limiting alcohol consumption to less than 100 grams per week or 1 drink daily for women and 2 drinks daily for men, avoiding binge drinking, and not initiating alcohol consumption as a preventive measure against ASCVD (1, 14).
Physical activity and exercise
Sedentary behavior is a well-established cardiovascular risk factor (15). Regular physical activity leads to improvements in measures of obesity, diabetes and insulin resistance, hypertension, and dyslipidemia, as well as enhancing endothelial function and reducing systemic inflammation; all of these collectively help reduce the risk of ASCVD (16, 17, 18, 19). Recommendations for all adults are to engage in at least 150 to 300 minutes of moderate-intensity or 75 to 150 minutes of vigorous-intensity physical activity weekly. For adults who are not able to perform these activities, the recommendation is to stay as active as their health condition allows (2). Resistance exercise for 2 or more days per week is also recommended (1).
Stress reduction and mental health care
Pharmacotherapy
Lipid lowering therapy
Statins primarily lower cardiovascular risk by inhibiting hepatic cholesterol synthesis through the inhibition of HMG-CoA reductase, which leads to the upregulation of liver LDL receptors and increased LDL clearance from the blood. Other potential beneficial effects of statins include enhanced endothelial nitric oxide production, stabilization of atherosclerotic plaques, reduced lipid accumulation in the arterial wall, and regression of plaques (24). However, statins may also carry risks, including myalgia and, rarely, rhabdomyolysis, as well as an increased risk of new-onset diabetes and potential liver enzyme elevations (25, 26).
Statin therapy is indicated for primary prevention of cardiovascular disease in the following groups (2):
Adults aged 20 to 75 years with high low density lipoprotein (LDL) cholesterol levels (≥ 190 mg/dL [≥ 4.9 mmol/L]): Maximally tolerated statin therapy
High-risk adults (estimated 10-year ASCVD risk: > 20%): High-intensity statin therapy
Adults with diabetes and multiple ASCVD risk factors: High-intensity statin therapy
Adults (aged 40 to 75 years) with diabetes, regardless of estimated 10-year ASCVD risk: Moderate-intensity statin therapy (consider high-intensity)
Intermediate-risk adults (estimated 10-year ASCVD risk: 7.5% to < 20%): Moderate-intensity statin therapy, following a discussion about risks
Intermediate-risk adults (estimated 10-year ASCVD risk: 7.5% to < 20%) and adults with borderline risks (estimated 10-year ASCVD risk 5% to < 7.5%) with a calcium score measurement:
Score of 0, and no diabetes, family history of premature ASCVD, cigarette smoking: Reasonable to withhold statin therapy and reassess in 5 to 10 years
Score of 1 to 99: Reasonable to initiate statin therapy for patients ≥ 55 years
Score of 100 or higher or 75th percentile or higher: Reasonable to initiate statin therapy
Intermediate-risk adults with risk-enhancing factors: Consider initiating or intensifying statin therapy
Borderline-risk adults with risk-enhancing factors: Consider moderate-intensity statin therapy
High-intensity statin therapy has the goal of lowering LDL cholesterol by ≥ 50%. Moderate-intensity statin therapy has the goal of lowering LDL cholesterol by 30 to 50%. Maximally tolerated statin therapy is the highest dose that is tolerated by the patient.
In patients with acute coronary syndrome, ischemic stroke, or established atherosclerotic cardiovascular disease (including peripheral artery disease), high-intensity statin therapy is recommended (27, 28, 29, 30). For patients already on maximally tolerated statin therapy who have an LDL cholesterol level of ≥ 70 mg/dL (≥ 1.8 mmol/L), a non-statin lipid-lowering agent is recommended. In addition, for this high-risk patient population, it is reasonable to further intensify lipid-lowering therapy if the LDL cholesterol level is 55 to 70 mg/dL (1.4 to 1.8 mmol/L) and the patient is already on a maximally tolerated statin therapy.
Ezetimibe lowers LDL cholesterol by blocking the uptake of cholesterol from the small intestine. When added to standard statin therapy, ezetimibe has been shown to reduce cardiovascular events in both patients with a prior acute coronary syndrome, and those with chronic coronary artery disease at very high risk, particularly when LDL cholesterol levels are ≥ 70 mg/dL ( ≥ 1.8 mmol/L) despite maximally tolerated statin therapy (29, 31).
Proprotein convertase subtilisin/kexin type 9 inhibitors, or PCSK9 inhibitors, are monoclonal antibodies (evolocumab, alirocumab) that target PCSK9. PCSK9 binds to LDL receptors on the surface of liver cells, promoting their degradation; the inhibition of PCSK9 leads to increased clearance of plasma LDL cholesterol. Clinical trials with evolocumab and alirocumab have shown reduction in atherosclerosis and cardiovascular events (32, 33). PCSK9 inhibitors are most frequently used in patients with severe primary hypercholesterolemia (LDL cholesterol ≥ 190 mg/dL [≥ 4.9 mmol/L]), with or without familial hypercholesterolemia, or in patients with established ASCVD for whom target LDL could not be achieved with maximally tolerated statin therapy. Inclisiran is a small interfering RNA (siRNA) therapeutic that also inhibits production of PCSK9 and has been shown to provide sustained LDL-lowering effects with infrequent (twice-yearly) dosing (34).
Other therapeutic agents, including small interfering RNAs (eg, olpasiran, lepodisiran) and antisense oligonucleotide technology (eg, pelacarsen), aim to specifically reduce lipoprotein(a) levels and are being evaluated for their efficacy and safety (35, 36).
Eicosapent ethyl is a highly purified form of eicosapentaenoic acid, a key omega-3 fatty acid. It reduces triglyceride levels by inhibiting liver triglyceride synthesis and enhancing the clearance of triglyceride-rich lipoproteins and has anti-inflammatory, endothelial stabilizing, and antiplatelet effects (37). It significantly reduces cardiovascular event rates in patients with cardiovascular disease who have elevated triglyceride levels despite statin therapy (38). Unlike prescription eicosapent ethyl, over-the-counter fish oil supplements typically contain a mixture of eicosapentaenoic acid and docosahexaenoic acid in lower and variable doses and have not consistently demonstrated cardiovascular event reduction in large clinical trials (39, 40).
Antiplatelet medications
Oral antiplatelet medications are essential in preventing atherosclerosis-related complications as most events originate from plaque fissure or rupture, leading to platelet activation and thrombosis. The following medications can be used:
Aspirin irreversibly inhibits cyclooxygenase-1 (COX-1) and disrupts thromboxane A2 production, inhibiting platelet activation and aggregation.
P2Y12 inhibitors (eg, clopidogrel, prasugrel, ticagrelor) block the adenosine diphosphate (ADP)-mediated activation of platelets.
Dual antiplatelet therapy with aspirin and an oral P2Y12 inhibitor is indicated for at least 12 months in patients with acute coronary syndrome who are not at high risk for bleeding (29). For individuals with low to moderate bleeding risk following acute coronary syndrome, it is recommended to switch from dual antiplatelet therapy to single antiplatelet therapy at 6 months following percutaneous coronary intervention (30).
Aspirin monotherapy at a low dose (75 to 100 mg) is recommended to reduce atherosclerotic events in patients with chronic coronary artery disease and no indication for oral anticoagulation (30). For patients with symptomatic peripheral artery disease, antiplatelet monotherapy with aspirin (75 to 325 mg daily) is also recommended to reduce the risk of adverse cardiovascular events (27). While aspirin therapy is a cornerstone of secondary prevention in patients with established cardiovascular disease, its role in primary prevention is controversial, and it is not recommended routinely for this purpose (2). It can be considered in patients 40 to 59 years of age with a 10-year ASCVD risk > 7.5% and low bleeding risk, but absolute benefit is likely to be small (41).
Single antiplatelet therapy with clopidogrel (75 mg daily) or aspirin (75 to 325 mg daily) is also recommended for preventing adverse events in patients with symptomatic peripheral artery disease (27).
Among the available P2Y12 inhibitors, clopidogrel is the least potent and takes the longest time to reach maximum platelet inhibition. While other P2Y12 inhibitors (prasugrel and ticagrelor) are more potent, they are also associated with higher bleeding risk. Therefore, in acute coronary syndrome, the use of clopidogrel is only recommended when other P2Y12 inhibitors are not available, not tolerated, or contraindicated (29). In individuals with a high risk of bleeding, de-escalation from dual antiplatelet therapy to single antiplatelet therapy with ticagrelor is recommended one month after percutaneous coronary intervention (29). The recommended duration of antiplatelet therapy also depends on concurrent anticoagulation. For example, no additional antiplatelet therapy is recommended in patients already on therapeutic anticoagulation who have no recent history of percutaneous revascularization or myocardial infarction.
Other medications
Beyond lipid-lowering agents and antiplatelet agents, additional medications are considered in patients with specific risk profiles or comorbidities. For example, sodium-glucose cotransporter 2 (SGLT2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists are used in patients with diabetes and heart failure, while anti-inflammatory therapies such as canakinumab or colchicine may benefit patients with persistent inflammatory risk after myocardial infarction. In selected patients with peripheral artery disease or after revascularization, low-dose anticoagulation combined with aspirin is recommended to further reduce ischemic events.
Thrombolysis remains a cornerstone for treatment of acute ischemic stroke (28). Thrombolysis involves administration of intravenous tissue plasminogen activator (tPA), such as alteplase, to dissolve occlusive thrombi that are obstructing cerebral blood flow. It is recommended for eligible patients who can be treated within 4.5 hours of the onset of stroke symptoms. Key indications include the diagnosis of acute ischemic stroke with measurable neurologic deficits, exclusion of intracranial hemorrhage by imaging, and careful consideration of contraindications such as recent surgery or gastrointestinal bleeding, concomitant use of anticoagulants, and severe uncontrolled hypertension. The primary goal is to restore cerebral perfusion, minimize brain injury, and improve clinical outcomes.
In acute coronary syndromes, the use of systemic thrombolysis is restricted to ST elevation myocardial infarction when the anticipated delay from first medical contact to primary percutaneous intervention is expected to be greater than 120 minutes (29).
Antihypertensives primarily affect atherosclerosis by reducing pressure and thus stress on the arterial wall. This effect is particularly important in aortic aneurysms, where the risk of rupture is directly related to blood pressure (42). For patients with thoracic aortic aneurysm, antihypertensive therapy is recommended with systolic blood pressure of ≥ 130 mm Hg or diastolic blood pressure of ≥ 80 mm Hg.
In the United States, for patients who are at low risk (< 7.5%% 10-year ASCVD risk based on the PREVENT calculator) with blood pressure is > 130/80, lifestyle intervention as an initial strategy followed by antihypertensive therapy if necessary (43). In patients with known coronary artery disease or whose risk of ASCVD is ≥ 7.5% as defined by PREVENT, antihypertensive medication is recommended as the initial therapy for blood pressure > 130/80 mm Hg.
By lowering glucose levels, some antihyperglycemic medications mitigate the harmful effects of hyperglycemia, which include endothelial dysfunction, increased oxidative stress, and chronic inflammation. The SGLT2 inhibitors reduce serum glucose by inhibiting renal glucose reabsorption, leading to glucosuria. SGLT2 inhibitors have demonstrated favorable effects on lipid metabolism, reduced inflammation, and improved endothelial function (44), and they have been associated with reduction in some major cardiovascular outcomes (45).
GLP-1 receptor agonists act by enhancing glucose-dependent insulin secretion, inhibiting glucagon secretion, and slowing gastric emptying. These agents may have substantial cardiovascular benefits, by preventing endothelial dysfunction through the promotion of angiogenesis and inhibition of oxidative stress; reducing systemic inflammation; and reducing monocyte recruitment, formation of proinflammatory macrophages and foam cells, vascular smooth muscle cell proliferation, and plaque development (46).
Canakinumab, an anti-inflammatory monoclonal antibody targeting interleukin-1beta, showed a significant reduction in cardiovascular events in patients with prior myocardial infarction and an elevated C-reactive protein level (47).
Colchicine, by inhibiting microtubule polymerization and consequent suppression of inflammatory cell activation and adhesion, has been shown to prevent major adverse cardiovascular events in patients with recent myocardial infarction and in patients with chronic coronary disease (but not in the setting of acute myocardial infarction) (48, 49).
Although routine anticoagulation is not generally advised for atherosclerosis treatment (or prevention), it can play a supplementary role in some cases. The combination of low-dose rivaroxaban (2.5 mg twice daily) and low-dose aspirin is recommended to reduce the risk of adverse cardiovascular and limb-related events in patients with symptomatic peripheral artery disease or following peripheral surgical or endovascular revascularization (27). Additionally, to reduce ischemic events in patients with ST elevation myocardial infarction treated with intravenous thrombolysis, parenteral anticoagulation is recommended to be continued for the entire hospital stay (up to 8 days) or until revascularization (29).
Catheter-based interventions
Catheter based interventions typically involve the displacement or removal of stenotic or occlusive atherosclerotic plaques. These interventions play an important role in the treatment of both acute complications of atherosclerosis and the management of hemodynamically significant chronic atherosclerotic lesions. In general, a lesion is considered hemodynamically significant when it results in impaired blood flow sufficient to cause symptoms or objective evidence of ischemia. While the exact thresholds vary by vascular territory, this often corresponds to luminal narrowing of ≥ 50% in coronary or carotid arteries when associated with clinical symptoms or imaging evidence of reduced perfusion and ≥ 70% for asymptomatic lesions. Functional assessments, such as fractional flow reserve (FFR) in the coronary circulation or pressure gradients and flow measurements in peripheral vessels, are also used to more accurately define hemodynamic significance. Treatment modalities include:
Balloon angioplasty
Stenting
Atherectomy
Intravascular lithotripsy
Thrombectomy
Brachytherapy
See also Percutaneous Coronary Interventions (PCI) for more information about catheter-based interventions in the setting of coronary artery disease.
Balloon angioplasty
Balloon angioplasty, also referred to as "plain-old balloon angioplasty" (POBA), is a percutaneous intervention involving advancement of a catheter with a deflated balloon to the site of arterial stenosis. The balloon is then inflated to compress atherosclerotic plaque materials against the vessel wall, thereby restoring luminal diameter and blood flow.
In addition to POBA, other techniques are available, including:
Drug-coated balloon angioplasty, which involves inflating a balloon that is coated with an antiproliferative medication such as paclitaxel or sirolimus. These medications help to prevent restenosis by inhibiting smooth muscle cell proliferation.
Cutting balloon angioplasty uses a balloon with micro blades on its surface to create controlled incisions in the plaque as the balloon is inflated. This technique is designed to treat resistant calcific lesions.
Stenting
Stent placement is frequently performed following balloon angioplasty. It involves the placement of an expandable metallic scaffold within an atherosclerotic artery to preserve luminal integrity and maintain adequate blood flow. Stenting can be performed by using:
Bare-metal stents are rarely used due to high restenosis rates and the superiority of drug-eluting stents.
Drug-eluting stents are most frequently used. These stents are coated with antiproliferative agents such as sirolimus, everolimus, paclitaxel, or zotarolimus to inhibit neointimal hyperplasia and reduce restenosis.
Covered stents are encased with a synthetic covering to seal aneurysms or manage other vascular abnormalities.
Biodegradable stents are designed to provide temporary scaffolding to maintain vessel patency and then gradually dissolve, potentially reducing long-term complications such as restenosis. Trials have shown mixed results in terms of outcomes compared to drug-eluting stents, so biodegradable stents are not standard of care (50).
Endovascular aneurysm repair (EVAR) is the insertion of a stent-graft via a catheter into the arterial lumen, followed by precise deployment within the aneurysm to strengthen the vessel wall and prevent aneurysmal rupture. EVAR is commonly used for the treatment of abdominal and thoracic aortic aneurysms (27).
In specific patient populations, such as those undergoing stenting of complex coronary lesions or left main disease, the use of intravascular imaging techniques such as intravascular ultrasound or optical coherence tomography is recommended for procedural guidance to optimize stent deployment and reduce future ischemic events (29).
Atherectomy
Atherectomy is an endovascular technique designed to excise or ablate atherosclerotic plaques from arterial walls, thereby restoring luminal patency and optimizing vascular flow. Potential techniques include:
Rotational atherectomy, which uses a high-speed, rotating diamond-coated burr to pulverize hard, calcific plaques.
Orbital atherectomy, which uses a diamond-coated crown that orbits within the artery, sanding down and pulverizing calcified plaques.
Laser atherectomy, which uses a laser-emitting catheter to vaporize plaque, converting it into small particles.
Intravascular lithotripsy
Intravascular lithotripsy uses acoustic shock waves to fracture and disrupt calcified atherosclerotic plaques. This technique is particularly effective in addressing heavily calcified plaques and is typically followed by balloon angioplasty and stent placement (51). It is primarily used in the treatment of coronary arteries and peripheral vessels (52).
Thrombectomy
Thrombectomy is a procedure used to remove intravascular thrombi to restore circulation. It can be performed using several techniques:
Mechanical thrombectomy: Mechanical removal of the thrombus using specialized devices
Aspiration thrombectomy: Aspiration of the thrombus using a suction device
Catheter-directed thrombolysis: Thrombolytic medications are delivered directly to the thrombus to dissolve it
Thrombectomy can be the preferred procedure in patients with acute stroke (28) or acute limb ischemia (27). However, in patients with acute coronary syndrome, the use of manual or aspiration thrombectomy is not recommended due to the lack of demonstrated benefit (29).
Brachytherapy
Brachytherapy delivers targeted radiation therapy within the arteries to prevent restenosis following angioplasty and stenting. Using a specialized catheter, radioactive isotopes are positioned at the site of the treated artery, where they emit localized radiation that inhibits vascular smooth muscle cell proliferation, thereby preventing neointimal hyperplasia. This localized radiation treatment is particularly beneficial for patients with recurrent in-stent restenosis (53).
Surgical interventions
Surgical techniques for atherosclerotic disease include:
Bypass surgery
Surgical endarterectomy
Aneurysm repair
Bypass surgery involves creating arterial connections to "bypass" blocked arteries using autogenous or prosthetic grafts, restoring blood flow to the affected area. Coronary artery bypass grafting (CABG) is critical in treating multivessel coronary artery disease (29, 53) and lower extremity peripheral artery disease (27). The decision to perform surgical or catheter-based revascularization in complex coronary artery disease should involve guideline-directed discussions, a team-based approach, and shared decision making. Patients with triple-vessel disease who have diabetes ideally should undergo surgical revascularization, with the left internal mammary artery (LIMA) grafted to the left anterior descending (LAD) artery, unless they are poor candidates for surgery, in which case percutaneous revascularization could be considered.
Surgical endarterectomy involves the excision of atherosclerotic plaque from the intimal layer of the artery, thereby restoring luminal diameter and improving blood flow. The most frequently performed variant is carotid endarterectomy, which is used to reduce ischemic stroke rate in patients with high-grade carotid stenosis. While recommendations vary, carotid endarterectomy is generally strongly recommended in patients with 70 to 99% symptomatic carotid stenosis (ie, those who have experienced a transient ischemic attack or stroke within the last 6 months corresponding to the vascular territory of the narrowed vessel) (54, 55). Early intervention is emphasized. Carotid endarterectomy may also be considered in patients with 50 to 69% symptomatic stenosis or in those with ≥ 60% asymptomatic stenosis, depending upon life expectancy and perioperative risk.
Aneurysm repair involves the surgical reinforcement of the weakened segment of an artery to prevent rupture. This is accomplished by replacing the aneurysmal vessel with a synthetic graft, which ensures vascular integrity. This procedure is primarily indicated for acute aortic complications or for large, high-risk abdominal and thoracic aortic aneurysms (39). The threshold for intervention varies based on several factors, including aneurysm size, location, growth rate, patient symptoms, and overall risk of rupture.
Key Points
The development of atherosclerosis involves the deposition of LDL particles, recruitment of inflammatory cells, endothelial dysfunction, and proliferation of smooth muscle cells, leading to the formation of fibrous and calcified plaques within the arterial wall.
Risk factors for atherosclerosis include age, family history of premature atherosclerosis, dyslipidemia, cardiovascular-kidney-metabolic factors (diabetes, hypertension, obesity and chronic kidney disease), inflammation, and lifestyle related risk factors (smoking, sedentary lifestyle, diet, alcohol consumption, and psychosocial factors).
Symptoms develop when plaque growth or rupture leads to reduced or obstructed blood flow, resulting in conditions such as angina, myocardial infarction, stroke, and claudication.
Diagnosis is typically confirmed through clinical evaluation and imaging techniques such as ultrasound, magnetic resonance imaging, or computed tomography.
Management includes lifestyle changes and medications such as lipid-lowering agents (statins, PCSK9 inhibitors, ezetimibe), antiplatelet medications (aspirin, P2Y12 inhibitors), antihypertensives, and antihyperglycemic agents (SGLT2 inhibitors, GLP-1 receptor agonists) to stabilize plaques and prevent cardiovascular events.
Catheter-based interventions include balloon angioplasty with or without stenting, atherectomy, intravascular lithotripsy, thrombectomy, and brachytherapy to mechanically manage and stabilize plaques or remove thrombi.
Surgical interventions aim to bypass (bypass grafting) or remove (endarterectomy) severe atherosclerotic lesions, while aneurysm repair with synthetic grafts is performed to prevent rupture.
Drug Information for the Topic





