



Pemphigus vulgaris is an uncommon, potentially fatal, autoimmune disorder characterized by intraepidermal blisters and extensive erosions on apparently healthy skin and mucous membranes. Diagnosis is by skin biopsy with direct and indirect immunofluorescence and enzyme-linked immunosorbent assay (ELISA) testing. Treatment is with corticosteroids and sometimes other immunosuppressive therapies.
Bullae are elevated, fluid-filled blisters ≥ 10 mm in diameter.
Pemphigus vulgaris usually occurs in middle-aged patients, affecting men and women in equal numbers. Rarely, cases have been reported in children. One variant, paraneoplastic pemphigus, can occur in patients who have malignant or benign tumors, most commonly non-Hodgkin lymphoma.
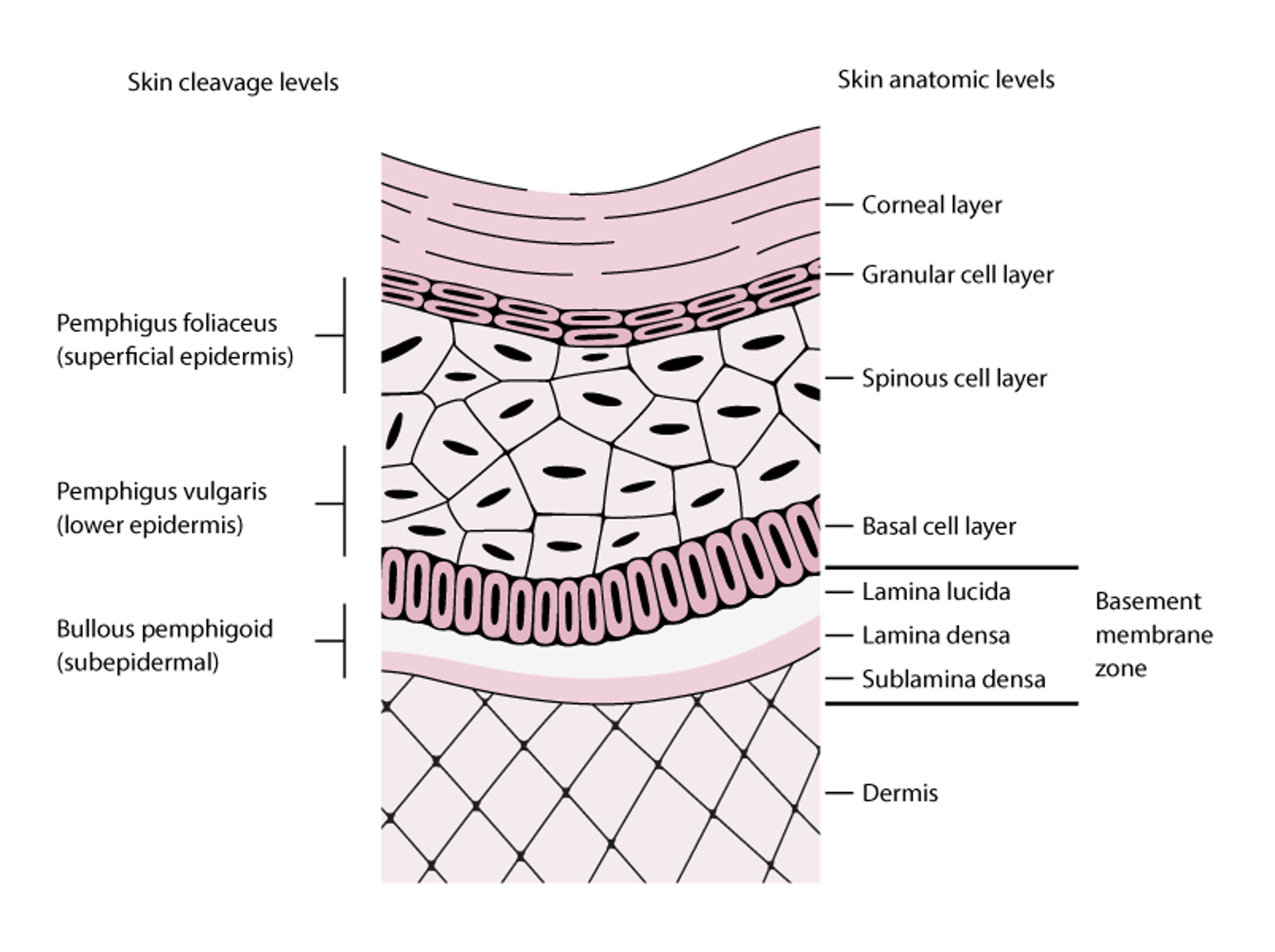
Pemphigus vulgaris is characterized by IgG autoantibodies directed against the calcium-dependent cadherins desmoglein 3 and sometimes desmoglein 1 (1). Paraneoplastic pemphigus has autoantibodies directed against these desmoglein antigens as well as others (eg, envoplakin, peiplakin, desmoplakin 1 and 2, BP-Ag 1). These transmembrane glycoproteins affect cell-cell adhesion and signaling between epidermal cells. Acantholysis (loss of intercellular adhesion with consequent epidermal blister formation) results from either direct inhibition of desmoglein function by autoantibody binding or from autoantibody-induced cell signaling that results in down-regulation of cell-cell adhesion. The autoantibodies are present in both serum and skin during active disease. Any area of stratified squamous epithelium may be affected, including mucosal surfaces (see figure Skin Cleavage Levels in Pemphigus and Bullous Pemphigoid).
Skin Cleavage Levels in Pemphigus and Bullous Pemphigoid
Pemphigus foliaceus blisters form in the superficial layers of the epidermis. Pemphigus vulgaris blisters can form at any epidermal level but typically form in the lower aspects of the epidermis. Bullous pemphigoid blisters form subepidermally (lamina lucida of the basement membrane zone). In this figure, the basement membrane zone is disproportionately enlarged to display its layers. |

Pemphigus (vulgaris, foliaceus, or both) may coexist with certain central nervous system (CNS) disorders, especially dementia, epilepsy, and Parkinson disease. Desmoglein 1 is present in CNS neurons (and in all epithelial cells), and an immunologic cross-reaction between epithelial and CNS isoforms has been suggested.
General reference
1. Russo I, De Siena FP, MD, Saponeri A, et al: Evaluation of anti-desmoglein-1 and anti-desmoglein-3 autoantibody titers in pemphigus patients at the time of the initial diagnosis and after clinical remission. Medicine (Baltimore) 96(46):e8801, 2017. doi: 10.1097/MD.0000000000008801
Symptoms and Signs of Pemphigus Vulgaris
Flaccid bullae, which are the primary lesions of pemphigus vulgaris, cause widespread and painful skin, oral, and other mucosal erosions. About half of patients have only oral erosions, which rupture and remain as chronic, painful lesions for variable periods. Often, oral lesions precede skin involvement. Dysphagia and poor oral intake are common because lesions also may occur in the upper esophagus. Cutaneous bullae typically arise in normal-appearing skin, rupture, and leave a raw area with crusting. Itching is usually absent. Erosions often become infected. If large portions of the body are affected, fluid and electrolyte loss may be significant.
A rare variant called pemphigus vegetans occurs primarily in intertriginous areas and oral mucosa with vegetating cauliflower-like plaques.

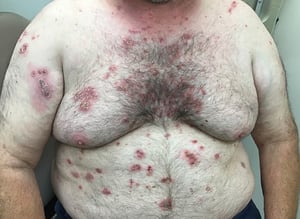
This photo shows generalized flaccid, ruptured bullae with surrounding erythema characteristic of pemphigus vulgaris.
This photo shows generalized flaccid, ruptured bullae with surrounding erythema characteristic of pemphigus vulgaris.
Photo courtesy of Daniel M. Peraza, MD.

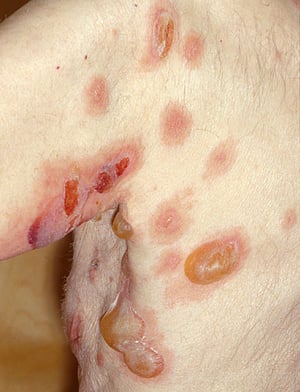
The primary lesions of pemphigus vulgaris are flaccid bullae.
The primary lesions of pemphigus vulgaris are flaccid bullae.
Photo provided by Thomas Habif, MD.

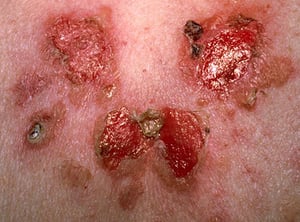
In pemphigus vulgaris, painful erosions on the skin and mucosa are common.
In pemphigus vulgaris, painful erosions on the skin and mucosa are common.
Photo provided by Thomas Habif, MD.

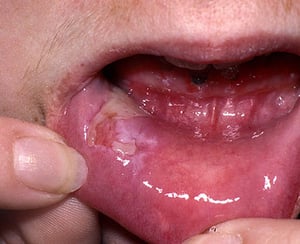
Pemphigus lesions typically appear first in the mouth, where they rupture and remain as chronic, painful erosions for variable periods. Oral lesions often precede cutaneous lesions.
Pemphigus lesions typically appear first in the mouth, where they rupture and remain as chronic, painful erosions for v
Photo provided by Thomas Habif, MD.

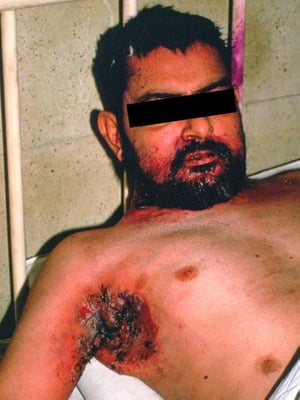
This photo shows erosions and fungating vegetating plaques in the axilla resulting from pemphigus vegetans.
This photo shows erosions and fungating vegetating plaques in the axilla resulting from pemphigus vegetans.
Photo courtesy of Karen McKoy, MD.
This photo shows generalized flaccid, ruptured bullae with surrounding erythema characteristic of pemphigus vulgaris.
This photo shows generalized flaccid, ruptured bullae with surrounding erythema characteristic of pemphigus vulgaris.
Photo courtesy of Daniel M. Peraza, MD.
The primary lesions of pemphigus vulgaris are flaccid bullae.
The primary lesions of pemphigus vulgaris are flaccid bullae.
Photo provided by Thomas Habif, MD.
In pemphigus vulgaris, painful erosions on the skin and mucosa are common.
In pemphigus vulgaris, painful erosions on the skin and mucosa are common.
Photo provided by Thomas Habif, MD.
Pemphigus lesions typically appear first in the mouth, where they rupture and remain as chronic, painful erosions for variable periods. Oral lesions often precede cutaneous lesions.
Pemphigus lesions typically appear first in the mouth, where they rupture and remain as chronic, painful erosions for v
Photo provided by Thomas Habif, MD.
This photo shows erosions and fungating vegetating plaques in the axilla resulting from pemphigus vegetans.
This photo shows erosions and fungating vegetating plaques in the axilla resulting from pemphigus vegetans.
Photo courtesy of Karen McKoy, MD.
Diagnosis of Pemphigus Vulgaris
Biopsy with immunofluorescence testing
Pemphigus vulgaris should be suspected in patients with unexplained chronic mucosal ulceration, particularly if they have bullous skin lesions. This disorder must be differentiated from other disorders that cause chronic oral ulcers and from other bullous dermatoses (eg, pemphigus foliaceus, bullous pemphigoid, mucous membrane pemphigoid, drug eruptions, toxic epidermal necrolysis, erythema multiforme, dermatitis herpetiformis, bullous contact dermatitis).
Two clinical findings, both reflecting lack of epidermal cohesion, that are somewhat specific for pemphigus vulgaris are the following:
Nikolsky sign (1): Upper layers of epidermis move laterally with slight pressure or rubbing of skin adjacent to a bulla.
Asboe-Hansen sign: Gentle pressure on intact bullae causes fluid to spread away from the site of pressure and beneath the adjacent skin.
The diagnosis of pemphigus vulgaris is confirmed by biopsy of lesional and surrounding (perilesional) normal skin. Immunofluorescence testing shows IgG autoantibodies against the keratinocyte's cell surface. Serum autoantibodies to desmoglein 1 and desmoglein 3 transmembrane glycoproteins can be identified via direct immunofluorescence, indirect immunofluorescence, and enzyme-linked immunosorbent assay (ELISA). Sensitivity of ELISA is higher (> 95% than that of indirect immunofluorescence (2).
Diagnosis references
1. Uzun S, Durdu M: The specificity and sensitivity of Nikolskiy sign in the diagnosis of pemphigus. J Am Acad Dermatol. 54(3):411-415, 2006. doi: 10.1016/j.jaad.2005.10.019
2. Joly P, Horvath B, Patsatsi Α, et al: Updated S2K guidelines on the management of pemphigus vulgaris and foliaceus initiated by the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol 34(9):1900-1913,2020. doi: 10.1111/jdv.16752
Treatment of Pemphigus Vulgaris
Corticosteroids, oral or IV
Sometimes immunosuppressants
Sometimes plasma exchange or IV immune globulin (IVIG)
Referral to a dermatologist with expertise in treating this disorder is recommended. Hospitalization is required initially for all but the most minor cases. Cleansing and dressing of open skin lesions is similar to that done to treat partial-thickness burns (eg, reverse isolation, hydrocolloid or silver sulfadiazine dressings).
Treatment of pemphigus vulgaris is aimed at decreasing production of pathogenic autoantibodies. One mainstay of treatment is systemic corticosteroids (1).
For mild pemphigus vulgaris, first-line treatment is typically rituximab (2) or prednisone (eg, 0.5 to 1 mg/kg once daily, depending on the extent of disease) with or without azathioprine or mycophenolate mofetil. Higher doses of prednisone may slightly hasten initial response but do not appear to improve outcome.
For moderate or severe pemphigus vulgaris, treatment is typically rituximab and higher doses of prednisone (eg, 1.0 to 1.5 mg/kg once daily) with or without azathioprine or mycophenolate mofetil. IV pulse therapy with methylprednisolone 1 g once a day also has been used.
The immunosuppressants rituximab (3), azathioprine, and mycophenolate mofetil help reduce the need for corticosteroids and thus minimize the undesirable effects of long-term corticosteroid use. Plasma exchange and high-dose IV immune globulin to reduce antibody titers have also been effective.
Treatment references
1. Joly P, Horvath B, Patsatsi Α, et al: Updated S2K guidelines on the management of pemphigus vulgaris and foliaceus initiated by the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol 34(9):1900-1913, 2020. doi: 10.1111/jdv.16752
2. Craythorne EE, Mufti G, DuVivier AW: Rituximab used as a first-line single agent in the treatment of pemphigus vulgaris. J Am Acad Dermatol 65(5):1064–1065, 2011. doi: 10.1016/j.jaad.2010.06.033
3. Joly P, Maho-Vaillant M, Prost-Squarcioni C, et al: First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): A prospective, multicentre, parallel-group, open-label randomised trial. Lancet 389(10083):2031–2040, 2017. doi: 10.1016/S0140-6736(17)30070-3
Prognosis for Pemphigus Vulgaris
Without treatment, pemphigus vulgaris is often fatal, usually within 5 years of disease onset. Systemic corticosteroid and immunosuppressive therapy has improved prognosis, but death may still result from complications of therapy.
Key Points
About half of patients with pemphigus vulgaris have only oral lesions.
Use Nikolsky and Asboe-Hansen signs to help clinically differentiate pemphigus vulgaris from other bullous disorders.
Confirm the diagnosis by immunofluorescence testing of skin samples.
Treat with systemic corticosteroids, with or without other immunosuppressive therapies (eg, rituximab), IV immune globulin, or plasma exchange.
Drug Information for the Topic





