



Bullous pemphigoid is a chronic autoimmune skin disorder resulting in generalized, pruritic, bullous lesions in older adults. Mucous membrane involvement is rare. Diagnosis is by skin biopsy and immunofluorescence testing of skin and serum. Topical and systemic corticosteroids are used initially. Most patients require long-term maintenance therapy, for which a variety of immunosuppressants can be used.
Bullae are elevated, fluid-filled blisters ≥ 10 mm in diameter.
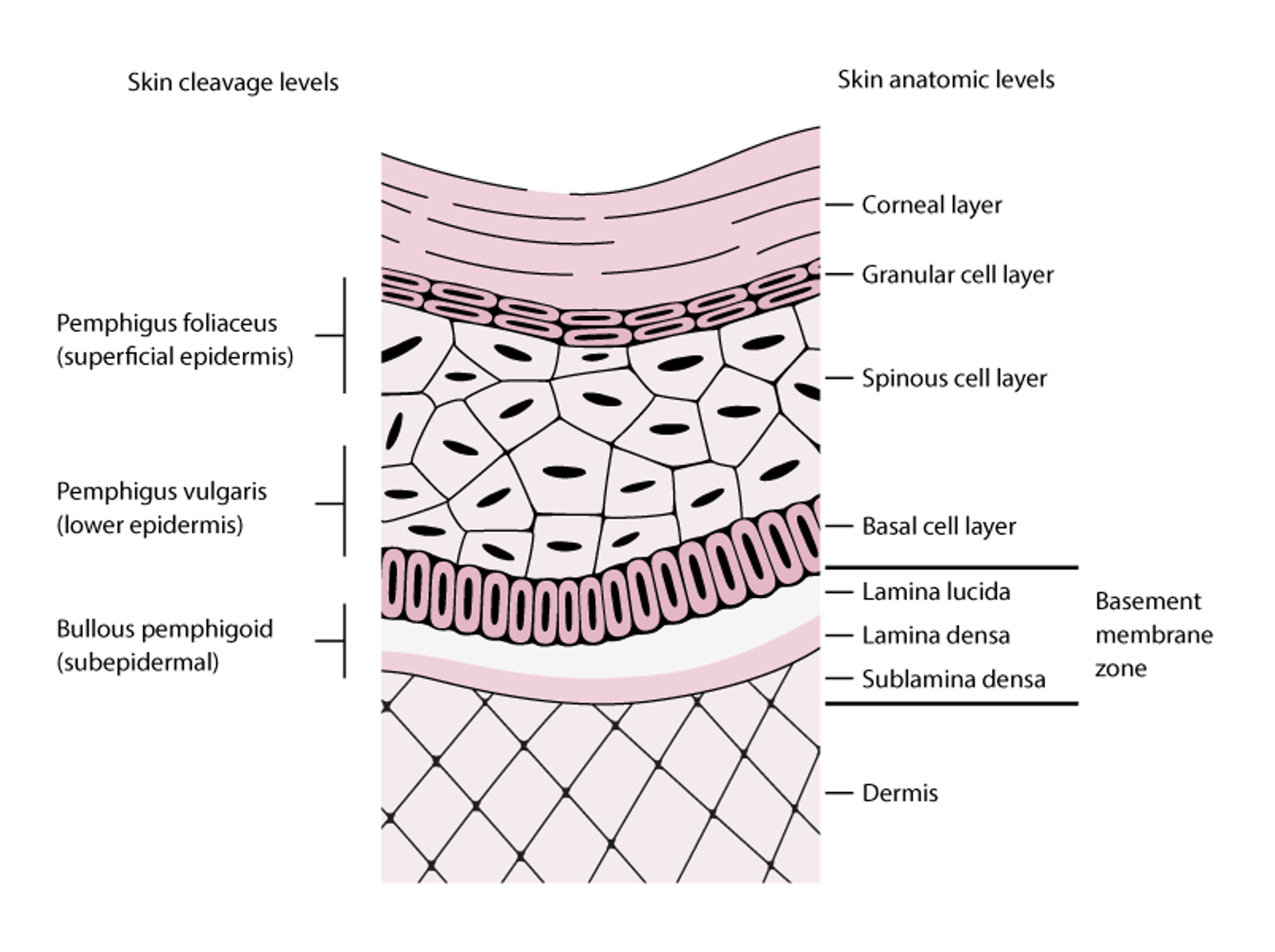
Bullous pemphigoid occurs more often in patients > age 60 but can occur in children. IgG autoantibodies bind to certain hemidesmosomal antigens (BPAg1 [BP230], BPAg2 [BP180]), resulting in the activation of complement to form a subepidermal blister (see figure Skin Cleavage Levels in Pemphigus and Bullous Pemphigoid).
Skin Cleavage Levels in Pemphigus and Bullous Pemphigoid
Pemphigus foliaceus blisters form in the superficial layers of the epidermis. Pemphigus vulgaris blisters can form at any epidermal level but typically form in the lower aspects of the epidermis. Bullous pemphigoid blisters form subepidermally (lamina lucida of the basement membrane zone). In this figure, the basement membrane zone is disproportionately enlarged to display its layers. |

Etiology of Bullous Pemphigoid
No cause of bullous pemphigoid has been proved; however, the following triggers have been suggested:
Drugs (including furosemide, spironolactone, omeprazole, PD-1 and PD-L1 monoclonal antibodies [eg, durvalumab, nivolumab, pembrolizumab], sulfasalazine, penicillin, penicillamine, etanercept, antipsychotics, and dipeptidyl peptidase-4 inhibitors)
Physical triggers (including trauma, radiation therapy for breast cancer, UV radiation, and anthralin)
Skin disorders (including psoriasis, lichen planus, and some infections)
Disorders (diabetes mellitus, rheumatoid arthritis, ulcerative colitis, and multiple sclerosis)
Genetic and environmental factors may play a role.
Triggers may induce an autoimmune reaction by mimicking molecular sequences in the epidermal basement membrane (molecular mimicry, as with drugs and possibly infections), by exposing or altering normally tolerated host antigens (as with physical triggers and certain disorders), or by other mechanisms. Epitope spreading refers to the recruitment of autoreactive lymphocytes against normally tolerated host antigens, which contributes to disease chronicity and course.
Certain central nervous system (CNS) and psychiatric disorders may precede bullous pemphigoid, especially multiple sclerosis and schizophrenia, but also dementias, intracranial bleeds, stroke, delusional and personality disorders, and Parkinson disease. To a lesser degree, these disorders may be preceded by bullous pemphigoid. Hypothesized shared causes include a cross-reactive immune response between neural and cutaneous antigens (BPAg1 is expressed in the CNS), as well as triggering by certain drugs used to treat the CNS disorders (eg, phenothiazine antipsychotics, spironolactone); however, a mechanism of triggering by drugs is not understood.
Symptoms and Signs of Bullous Pemphigoid
Pruritus is the first symptom of bullous pemphigoid. Skin lesions may not develop for several years. Often, characteristic tense bullae develop on skin of the trunk and in the flexural and intertriginous areas. Bullae may develop on normal-appearing skin or may be preceded by erythematous or urticarial-appearing plaques. Localized disease may occur at trauma sites, stomas, and anogenital and lower leg areas. Dyshidrotic pemphigoid is a rare form of bullous pemphigoid that affects the hands and feet and can look like dyshidrotic dermatitis (a form of hand and foot dermatitis) on the palms. Bullae usually do not rupture, but those that do often rapidly heal.
Polymorphic, annular, dusky-red, edematous lesions, with or without peripheral vesicles, can occur. Rarely, small blisters develop on the mucosa. Leukocytosis and eosinophilia are common, but fever is rare. The Nikolsky sign, where upper layers of epidermis move laterally with slight pressure or rubbing of skin adjacent to a blister, is negative.

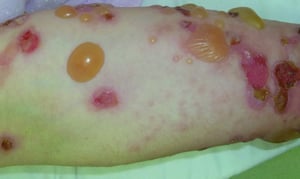
This photo shows tense bullae, erosions, and crusts on the arm of a patient with bullous pemphigoid.
This photo shows tense bullae, erosions, and crusts on the arm of a patient with bullous pemphigoid.
© Springer Science+Business Media

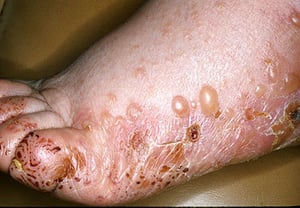
This photo of a patient with bullous pemphigoid shows erythematous plaques with intact bullae and erosions on the lateral and dorsal foot.
This photo of a patient with bullous pemphigoid shows erythematous plaques with intact bullae and erosions on the later
Photo provided by Thomas Habif, MD.

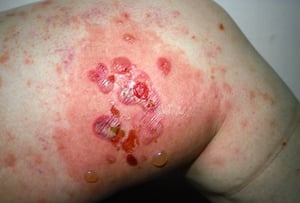
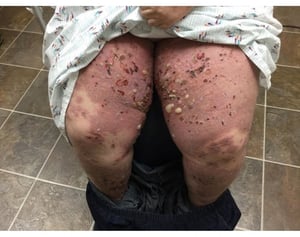
This photo shows ruptured bullae and erosions on the thigh of a person with bullous pemphigoid.
This photo shows ruptured bullae and erosions on the thigh of a person with bullous pemphigoid.
Photo courtesy of Karen McKoy, MD.

This photo shows ruptured and unruptured bullous lesions on an erythematous base.
This photo shows ruptured and unruptured bullous lesions on an erythematous base.
Photo courtesy of Daniel M. Peraza, MD.

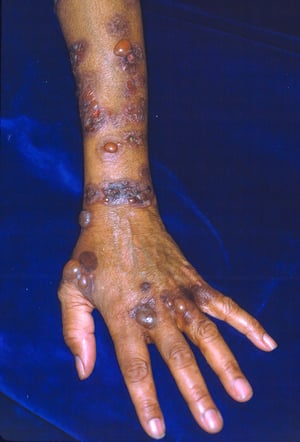
This photo shows tense bullae, erosions, and hyperpigmentation on the arm of a patient with bullous pemphigoid.
This photo shows tense bullae, erosions, and hyperpigmentation on the arm of a patient with bullous pemphigoid.
Photo courtesy of Karen McKoy, MD.
This photo shows tense bullae, erosions, and crusts on the arm of a patient with bullous pemphigoid.
This photo shows tense bullae, erosions, and crusts on the arm of a patient with bullous pemphigoid.
© Springer Science+Business Media
This photo of a patient with bullous pemphigoid shows erythematous plaques with intact bullae and erosions on the lateral and dorsal foot.
This photo of a patient with bullous pemphigoid shows erythematous plaques with intact bullae and erosions on the later
Photo provided by Thomas Habif, MD.
This photo shows ruptured bullae and erosions on the thigh of a person with bullous pemphigoid.
This photo shows ruptured bullae and erosions on the thigh of a person with bullous pemphigoid.
Photo courtesy of Karen McKoy, MD.
This photo shows ruptured and unruptured bullous lesions on an erythematous base.
This photo shows ruptured and unruptured bullous lesions on an erythematous base.
Photo courtesy of Daniel M. Peraza, MD.
This photo shows tense bullae, erosions, and hyperpigmentation on the arm of a patient with bullous pemphigoid.
This photo shows tense bullae, erosions, and hyperpigmentation on the arm of a patient with bullous pemphigoid.
Photo courtesy of Karen McKoy, MD.
Diagnosis of Bullous Pemphigoid
Skin biopsy and IgG titers
If blisters develop, bullous pemphigoid needs to be differentiated from pemphigus vulgaris, a blistering disorder with a worse prognosis; differentiation is usually possible using clinical criteria.
Distinguishing Bullous Pemphigoid From Pemphigus Vulgaris
Disorder | Appearance of Lesion | Oral Involvement | Itching | Nikolsky Sign | Prognosis |
|---|---|---|---|---|---|
Bullous pemphigoid | Tense bullae that develop on normal-appearing or erythematous skin or urticarial-appearing plaques | Rare, with small blisters | Common | Usually negative | Usually good; occasionally fatal in older adults |
Flaccid bullae of various sizes Often shearing off of skin or mucosa, leaving painful erosions | Typically starts in the mouth | Absent | Positive | Mortality ≤ 10% with treatment; higher without treatment |
Test results help differentiate bullous pemphigoid from pemphigus vulgaris, linear IgA disease, erythema multiforme, drug-induced eruptions, mucous membrane pemphigoid, paraneoplastic pemphigoid, dermatitis herpetiformis, and epidermolysis bullosa acquisita.
If bullous pemphigoid is suspected, skin biopsy is done for histology and direct immunofluorescence testing. Samples from in and around the lesion itself are often used for histology, but samples of uninvolved skin (often about 3 mm from the edge of a lesion) are used for direct immunofluorescence. The blister in bullous pemphigoid is subepidermal, often containing many neutrophils and eosinophils. Direct immunofluorescence shows linear IgG and complement deposits along the basement membrane zone (dermal–epidermal junction). Indirect immunofluorescence shows circulating IgG deposits on the epidermal side of a salt-split preparation of normal (ie, test substrate) skin.
Serum is tested for IgG antibodies to BPAg1 and BPAg2 using an enzyme-linked immunosorbent assay (ELISA). Circulating IgG autoantibodies are present in about three fourths of patients (1). Newer, potentially more accurate ELISA tests are under investigation.
Diagnosis references
1. van Beek N, Krüger S, Fuhrmann T, et al: Multicenter prospective study on multivariant diagnostics of autoimmune bullous dermatoses using the BIOCHIP technology. J Am Acad Dermatol 83(5):1315-1322, 2020. doi: 10.1016/j.jaad.2020.01.049
Treatment of Bullous Pemphigoid
Corticosteroids, topical or oral
Anti-inflammatory drugs
Immunosuppressant drugs
High-potency topical corticosteroids (eg, clobetasol 0.05% cream) should be used for localized disease and may reduce the required dose of systemic drugs (1).
Patients with generalized disease often require systemic treatment with prednisone 0.5 mg/kg orally once a day, which can be tapered to a maintenance level of ≤ 0.1 mg/kg/day after several weeks. Most patients achieve remission after 2 to 10 months, but treatment may need to continue for several years before the disease process remits enough to allow discontinuation. If long-term therapy is necessary, a new blister every few weeks does not require increasing the prednisone dose.
Bullous pemphigoid occasionally responds to the anti-inflammatory activity of certain drugs, such as the combination of tetracycline or minocycline and nicotinamide. Other treatment options include monotherapy with dapsone, sulfapyridine, or erythromycin. IV immune globulin has been used occasionally.
For patients with generalized and recalcitrant disease, and sometimes to decrease corticosteroid dose in chronic disease, immunosuppressants such as methotrexate, azathioprine, mycophenolate mofetil, and cyclosporine may be used. Among the biologics, rituximab, dupilumab, omalizumab, and intravenous immunoglobulins may be used.
Treatment reference
1. Borradori L, Van Beek N, Feliciani C, et al: Updated S2 K guidelines for the management of bullous pemphigoid initiated by the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol 36(10):1689-1704, 2022. doi: 10.1111/jdv.18220
Prognosis for Bullous Pemphigoid
Bullous pemphigoid is a chronic disease. Although topical and systemic therapies are helpful, they may cause adverse effects.
Remission is typical within months, but treatment is sometimes needed for several years.
Key Points
Bullous pemphigoid usually affects patients > age 60 and is autoimmune and idiopathic.
Pruritus may precede development of a rash by years, and mucous membrane involvement is rare.
Biopsy the skin for histology and immunofluorescence testing and measure circulating autoantibodies.
Treat patients with high-potency topical corticosteroids when possible to avoid or minimize use of systemic corticosteroids.
Anti-inflammatory and immunosuppressant therapy may be used to limit corticosteroid dose.
Symptoms usually lessen within months, but treatment is sometimes needed for several years.
Drug Information for the Topic





