



Systemic sclerosis is a rare, chronic systemic rheumatic disease of unknown cause characterized by diffuse fibrosis and vascular abnormalities in the skin, joints, and internal organs (especially the esophagus, lower gastrointestinal tract, lungs, heart, and kidneys). Common symptoms include Raynaud syndrome, polyarthralgia, dysphagia, heartburn, and swelling and eventually skin thickening and contractures of the fingers. Lung, heart, and kidney involvement accounts for most deaths. Diagnosis is clinical, but laboratory tests support the diagnosis and aid in prognostication. Specific treatment is based on symptoms and specific organ involvement.
Systemic sclerosis is more common among women than men, with one systematic review citing a 5-fold higher prevalence among women (1). It is most common among people aged 20 to 50 and is rare in children.
Reference
1. Bairkdar M, Rossides M, Westerlind H, Hesselstrand R, Arkema EV, Holmqvist M. Incidence and prevalence of systemic sclerosis globally: a comprehensive systematic review and meta-analysis. Rheumatology (Oxford). 2021;60(7):3121-3133. doi:10.1093/rheumatology/keab190
Classification of Systemic Sclerosis
Systemic sclerosis is classified as
Limited cutaneous systemic sclerosis (CREST syndrome)
Diffuse cutaneous systemic sclerosis
Systemic sclerosis sine scleroderma
In limited cutaneous systemic sclerosis (CREST syndrome—calcinosis cutis, Raynaud syndrome, esophageal dysmotility, sclerodactyly, telangiectasias), patients develop skin thickening over the face and distal to the elbows and knees and may also have gastroesophageal reflux disease. This type is characterized by slow progression and is often complicated by pulmonary hypertension.
In diffuse cutaneous systemic sclerosis, patients have skin involvement that extends more proximally than in patients with limited cutaneous disease. They also have Raynaud syndrome and gastrointestinal (GI) complications. This subset may evolve rapidly. Interstitial lung disease and scleroderma renal crisis are the major complications.
In systemic sclerosis sine scleroderma, patients have systemic sclerosis–related antibodies and visceral manifestations of the disease but no skin thickening.
Etiology of Systemic Sclerosis
Immunologic mechanisms and heredity play a role in etiology.
Systemic sclerosis–like syndromes have been associated with vinyl chloride, bleomycin, pentazocine, epoxy and aromatic hydrocarbons, contaminated rapeseed oil, or L-tryptophan.
Pathophysiology of Systemic Sclerosis
Pathophysiology involves vascular damage and activation of fibroblasts; collagen and other extracellular proteins in various tissues are overproduced.
In systemic sclerosis, the skin develops more compact collagen fibers in the reticular dermis, epidermal thinning, loss of rete pegs (epithelial extensions that project into the underlying connective tissue), and atrophy of dermal appendages. T cells may accumulate, and extensive fibrosis in the dermal and subcutaneous layers develops. In the nail folds, capillary loops dilate, and some microvascular loops are lost. In the extremities, chronic inflammation and fibrosis of the synovial membrane and surfaces and periarticular soft tissues occur.
Esophageal motility becomes impaired, and the lower esophageal sphincter becomes incompetent; gastroesophageal reflux and secondary strictures can develop. The intestinal muscularis mucosa degenerates, leading to pseudodiverticula in the colon and ileum. Interstitial and peribronchial fibrosis or intimal hyperplasia of small pulmonary arteries can develop; if long-standing, pulmonary hypertension can result. Diffuse myocardial fibrosis or cardiac conduction abnormalities occur. Intimal hyperplasia of interlobular and arcuate arteries can develop within the kidneys, causing renal ischemia and hypertension.
Systemic sclerosis varies in severity and progression, ranging from generalized skin thickening with rapidly progressive and often fatal visceral involvement (diffuse cutaneous systemic sclerosis) to isolated skin involvement (often just the fingers and face) and slow progression (often several decades). The latter form is termed limited cutaneous systemic sclerosis including CREST syndrome. In addition, systemic sclerosis can overlap with other systemic rheumatic diseases—eg, sclerodermatomyositis (tight skin and muscle weakness indistinguishable from idiopathic inflammatory myopathy) and mixed connective tissue disease.
Symptoms and Signs of Systemic Sclerosis
The most common initial symptoms and signs of systemic sclerosis are Raynaud syndrome and insidious swelling of the distal extremities (puffy fingers) with gradual thickening of the skin of the fingers. Polyarthralgia is also prominent. Gastrointestinal disturbances (eg, heartburn, dysphagia) or respiratory complaints (eg, dyspnea) are occasionally the first manifestations.
Skin and nail manifestations
Swelling of the skin is usually symmetric and progresses to induration. It may be confined to the fingers (sclerodactyly) and hands, or it may affect most or all of the body. The skin eventually becomes taut, shiny, and hypopigmented or hyperpigmented; the face becomes masklike, with a small, pinched nose and a fish mouth appearance. Telangiectases may appear on the fingers, chest, face, lips, and tongue. However, in some patients, skin can then soften to variable degrees after a few years.
Subcutaneous calcifications may develop, usually on the fingertips (pulps) and over bony eminences.
Digital ulcers are common, especially on the fingertips, overlying the finger joints, or over calcinotic nodules.
Gingival and periodontal diseases are common, as well as alopecia.
Abnormal capillary and microvascular loops in the nails can be seen with an ophthalmoscope or dissecting microscope.

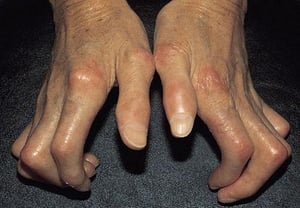
This photo shows late-stage shiny and thickened skin with effacement of normal markings secondary to tautness, called sclerodactyly.
This photo shows late-stage shiny and thickened skin with effacement of normal markings secondary to tautness, called s
By permission of the publisher. From Pandya A: Gastroenterology and Hepatology: Stomach and Duodenum. Edited by M Feldman. Philadelphia, Current Medicine, 1996.

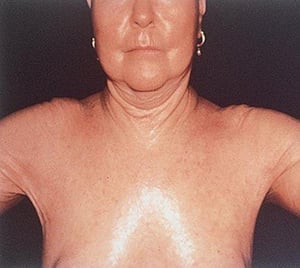
In this photo, hidebound, taut skin throughout the chest also extends over the shoulders bilaterally, leading to a loss of range of movement of the shoulders.
In this photo, hidebound, taut skin throughout the chest also extends over the shoulders bilaterally, leading to a loss
By permission of the publisher. From Marder W, Lath V, Crofford L, Lowe L, McCune WJ: Atlas of Rheumatology. Edited by G Hunder. Philadelphia, Current Medicine, 2005.

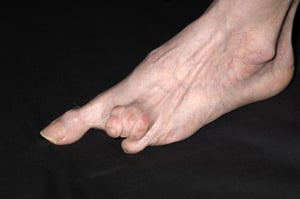
In this photo, hardening and tightening of the skin has caused toes to curl in on themselves.
In this photo, hardening and tightening of the skin has caused toes to curl in on themselves.
DR P. MARAZZI/SCIENCE PHOTO LIBRARY


This photo shows tight, firm, shiny skin on the forearm of a patient with systemic sclerosis.
This photo shows tight, firm, shiny skin on the forearm of a patient with systemic sclerosis.
Photo courtesy of Karen McKoy, MD.
This photo shows late-stage shiny and thickened skin with effacement of normal markings secondary to tautness, called sclerodactyly.
This photo shows late-stage shiny and thickened skin with effacement of normal markings secondary to tautness, called s
By permission of the publisher. From Pandya A: Gastroenterology and Hepatology: Stomach and Duodenum. Edited by M Feldman. Philadelphia, Current Medicine, 1996.
In this photo, hidebound, taut skin throughout the chest also extends over the shoulders bilaterally, leading to a loss of range of movement of the shoulders.
In this photo, hidebound, taut skin throughout the chest also extends over the shoulders bilaterally, leading to a loss
By permission of the publisher. From Marder W, Lath V, Crofford L, Lowe L, McCune WJ: Atlas of Rheumatology. Edited by G Hunder. Philadelphia, Current Medicine, 2005.
In this photo, hardening and tightening of the skin has caused toes to curl in on themselves.
In this photo, hardening and tightening of the skin has caused toes to curl in on themselves.
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
This photo shows tight, firm, shiny skin on the forearm of a patient with systemic sclerosis.
This photo shows tight, firm, shiny skin on the forearm of a patient with systemic sclerosis.
Photo courtesy of Karen McKoy, MD.
Joint manifestations

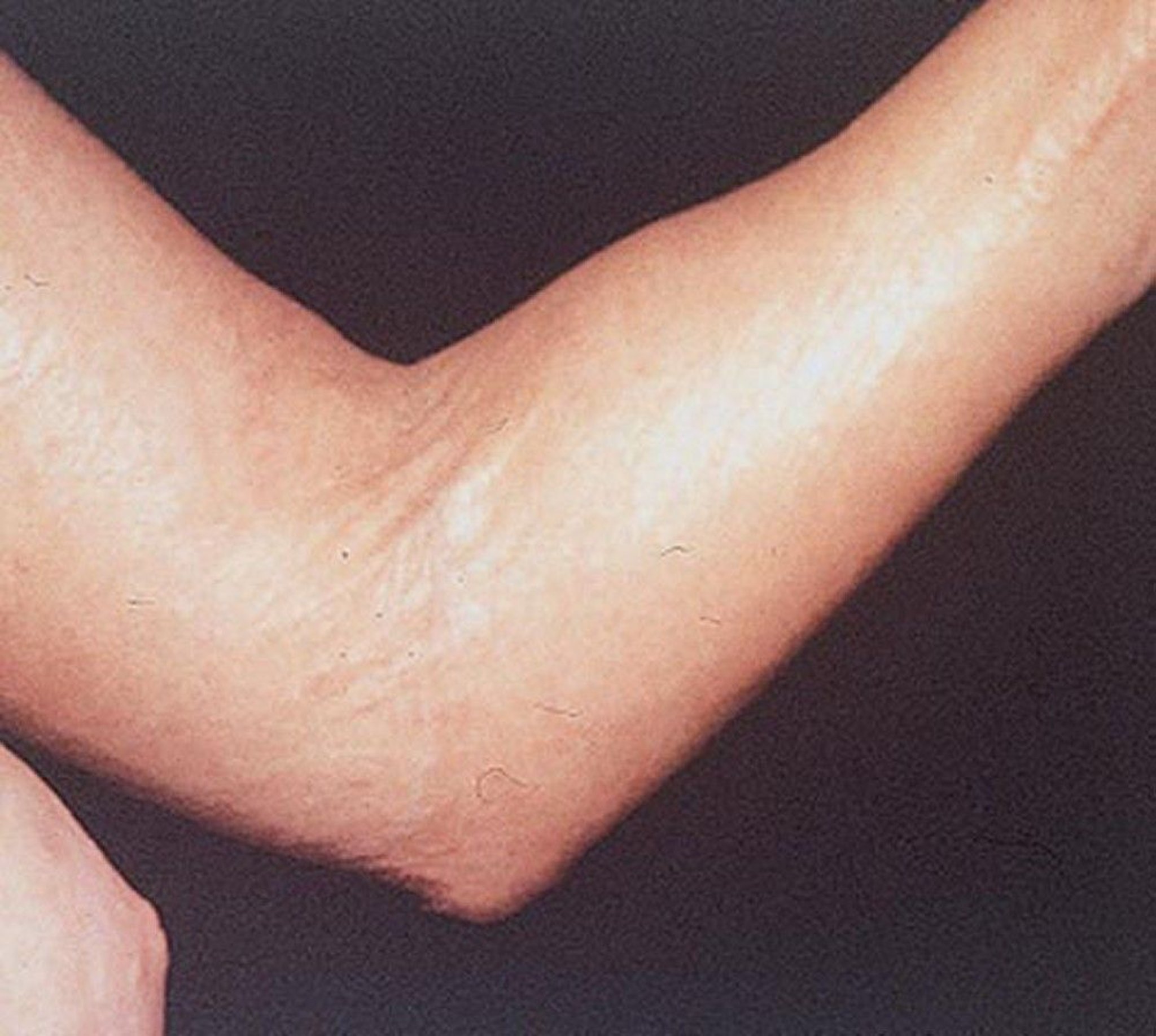
Flexion contracture of the elbow is seen in this photo of a patient with diffuse cutaneous systemic sclerosis.
By permission of the publisher. From Marder W, Lath V, Crofford L, Lowe L, McCune WJ: Atlas of Rheumatology. Edited by G Hunder. Philadelphia, Current Medicine, 2005.
Polyarthralgias or mild arthritis can be prominent. Flexion contractures may develop in the fingers, wrists, and elbows.
Friction rubs may develop over the joints, tendon sheaths, and large bursae.
Gastrointestinal manifestations
Esophageal dysfunction is the most frequent visceral disturbance and occurs in most patients. Dysphagia (usually retrosternal) usually develops first. Acid reflux can cause heartburn and stricture. Barrett esophagus occurs in one-third of patients and predisposes to complications (eg, adenocarcinoma).
Hypomotility of the small bowel causes bowel pseudo-obstruction and bacterial overgrowth that can lead to malabsorption. Air may penetrate the damaged bowel wall and be visible on radiographs (pneumatosis intestinalis). Leakage of bowel contents into the peritoneal cavity can cause peritonitis. Distinctive wide-mouthed pseudodiverticula can develop in the colon.
Primary biliary cholangitis may occur in patients with limited cutaneous systemic sclerosis (CREST syndrome).
Cardiopulmonary manifestations
Lung involvement generally progresses indolently, with substantial individual variability, but is a common cause of death. Lung fibrosis and interstitial lung disease are common and can impair gas exchange, leading to exertional dyspnea and restrictive disease with eventual respiratory failure. Acute alveolitis (potentially responsive to therapy) can develop. Esophageal dysfunction can lead to aspiration pneumonia.
Pulmonary hypertension may develop, as can heart failure, both of which are poor prognostic findings. Pericarditis with effusion or pleurisy can occur. Cardiac arrhythmias are common.
Renal manifestations
Severe, often sudden-onset renal disease (scleroderma renal crisis) may occur, most commonly in the first 4 to 5 years in patients who usually have diffuse cutaneous scleroderma and the RNA polymerase III antibody. It is often heralded by sudden, severe hypertension with features of thrombotic microangiopathic hemolytic anemia. It can also occur without acute hypertension or in systemic sclerosis sine scleroderma, and therefore clinical suspicion is required to make the diagnosis.
Corticosteroid use is a risk factor for development of scleroderma renal crisis.
Diagnosis of Systemic Sclerosis
Antibody testing
Clinical criteria
Systemic sclerosis should be considered in patients with Raynaud syndrome, typical musculoskeletal or skin manifestations, or unexplained dysphagia, malabsorption, pulmonary fibrosis, pulmonary hypertension, or cardiomyopathies. Diagnosis of systemic sclerosis sine scleroderma should be considered in patients who have unexplained visceral findings (eg, pulmonary hypertension). Diagnosis of systemic sclerosis can be obvious in patients with a combination of classic manifestations, such as Raynaud syndrome (with abnormal nail-fold capillary findings), dysphagia, and thickened skin. However, in some patients, the diagnosis cannot be made clinically; confirmatory laboratory tests can increase the probability of disease, but the absence of some laboratory and physical findings does not exclude it.
Antibody testing
The most cost-effective way to test for antibodies is to test for antinuclear antibodies (ANA), anti-Scl-70, anticentromere antibodies, and anti-U3 RNP antibodies first; if results are negative, testing for other antibodies should be considered based on clinical manifestations.
ANA are present in ≥ 90% of patients, often with an antinucleolar pattern.
Anticentromere antibodies occur in the serum of a high proportion of patients with limited cutaneous disease but are not specific.
Anti-Scl-70 (topoisomerase I) antibodies are more likely in patients with diffuse cutaneous systemic sclerosis than in those with limited cutaneous disease.
Anti-RNA polymerase III antibody is associated with diffuse cutaneous systemic sclerosis, scleroderma renal crisis, and cancer.
Anti-U3 RNP (fibrillarin) antibody is also associated with diffuse disease.
Anti-Th/To antibodies are associated with limited disease, interstitial lung disease, and pulmonary arterial hypertension.
Anti-PM-Scl antibodies may be present in patients with myositis, calcinosis, and interstitial lung disease and have a more favorable prognosis (1).
Clinical criteria
To help establish the diagnosis, clinicians can also consult the American College of Rheumatology (ACR)/European League Against Rheumatism's (EULAR) classification criteria for systemic sclerosis.
ACR/EULAR criteria for systemic sclerosis include the following features:
Skin thickening of the fingers of both hands
Fingertip lesions (eg, ulcers, pitting scars)
Telangiectasia
Abnormal nail-fold capillaries (eg, ectatic blood vessels, dropout areas) on capillaroscopy examination (eg, seen with an ophthalmoscope or dissecting microscope)
Pulmonary arterial hypertension and/or interstitial lung disease
Raynaud phenomenon
Systemic sclerosis–related autoantibodies (anticentromere, anti-Scl-70, anti-RNA polymerase III)
These criteria are weighted, in some cases according to subcriteria, and added to generate a score. Scores above a certain threshold are classified as definite systemic sclerosis.
As part of baseline evaluation, pulmonary function testing, high-resolution chest CT (with supine and prone position to ensure that early changes are not due to atelectasis), and echocardiography are used to document cardiopulmonary involvement (interstitial lung disease and/or pulmonary hypertension) and severity of disease. The initial evaluation is indicated even in patients who do not report dyspnea, cough, or exercise intolerance. Echocardiography and pulmonary function testing should be done every 1 to 2 years thereafter.

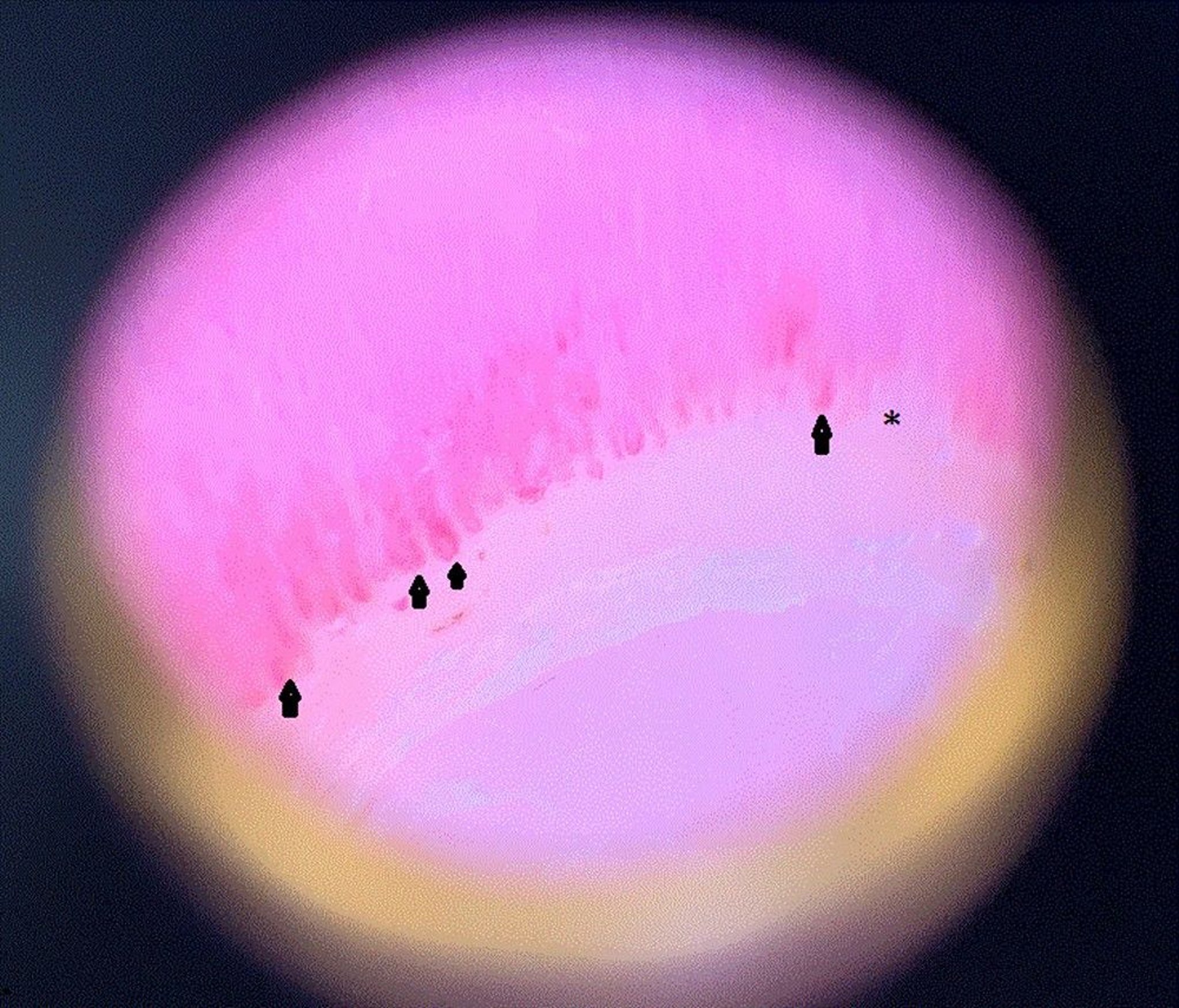
This image shows dilated capillary loops (arrowheads) in the nail fold of a patient with systemic sclerosis. Also seen is a dropout area (asterisk), which means the capillary ends prematurely and drops out and therefore looks shorter than the others.
Image courtesy of Sanjeev Patil, MD.

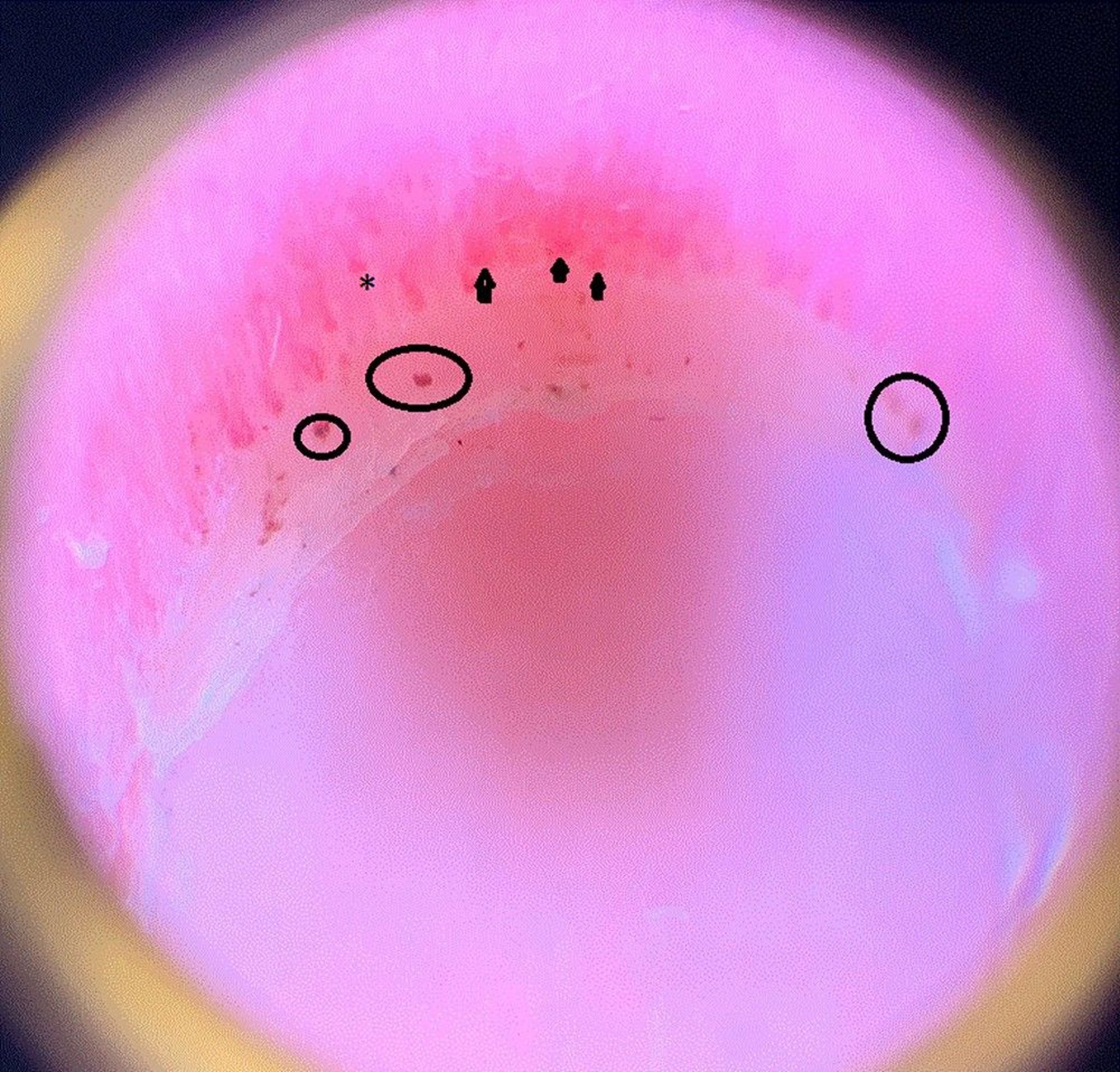
This image shows dilated capillary loops (arrowheads); a dropout area, which means the capillary ends prematurely and drops out and therefore looks shorter than the others (asterisk); and periungual hemorrhages (circles) in the nail fold of a patient with systemic sclerosis.
Image courtesy of Sanjeev Patil, MD.
Diagnosis reference
1. Lazzaroni MG, Marasco E, Campochiaro C, et al. The clinical phenotype of systemic sclerosis patients with anti-PM/Scl antibodies: results from the EUSTAR cohort. Rheumatology (Oxford). 2021;60(11):5028-5041. doi:10.1093/rheumatology/keab152
Treatment of Systemic Sclerosis
Treatment directed at symptoms and affected organs
No single medication significantly influences the natural course of systemic sclerosis overall, but various medications are of value in treating specific symptoms or involved organ systems.
Corticosteroids predispose patients to scleroderma renal crisis and thus should be avoided (1).
Skin and joint manifestations
No definitive treatment has been shown to help with skin involvement. However, methotrexate, mycophenolate mofetil, tocilizumab, rituximab, IV immune globulin, and cyclophosphamide all have shown modest benefit (2).
No treatment has clear benefit in treating calcinosis. Several agents, such as warfarin, hydroxychloroquine, heparin, and colchicine, have been studied but have not yielded promising results. However, sodium thiosulfate injected into a lesion can be used, as well as surgical excision of large lesions that affect joint function or that are cosmetically distressing for the patient.
Laser therapy might be helpful for telangiectasias that are cosmetically distressing for the patient, but the recurrence rate is high.
There is evidence that autologous hematopoietic stem cell in early diffuse cutaneous systemic sclerosis increases survival after the first year more than does IV cyclophosphamide; however, mortality was higher during the first year (3). The high incidence of mortality has limited its use, which is generally limited to specialized centers.
Physical therapy may help preserve muscle strength, but is ineffective in preventing joint contractures.
Gastrointestinal manifestations
Reflux esophagitis is relieved by frequent small feedings, high-dose proton pump inhibitors, sleeping with the head of the bed elevated, and not lying supine within 3 hours of the last meal.
Esophageal strictures may require periodic dilation; gastroesophageal reflux may possibly require gastroplasty.
Patients may require treatment with antibiotics to address symptoms of small intestine bacterial overgrowth, such as bloating, flatulence, and diarrhea.
Cardiopulmonary manifestations
Mycophenolate mofetil is effective for the treatment of scleroderma-associated interstitial lung disease (4). It was compared to cyclophosphamide and was found to be as effective and better tolerated (4).
The anti-IL-6 blocker tocilizumab has been shown to preserve lung function in scleroderma-associated interstitial lung disease and is becoming an alternative to mycophenolate mofetil, especially if polyarthritis is a predominant feature (5).
The antifibrotic agent nintedanib has been shown to slow the rate of lung function decline in scleroderma-associated interstitial lung disease and can be added to immunosuppressive therapy in patients whose disease progresses despite adequate immunosuppressive treatment (6).
Rituximab is being evaluated for connective tissue disease–related interstitial lung disease and may have efficacy (7). Azathioprine also may help with interstitial lung disease.
Successful lung transplantation also has been reported (8).
Epoprostenol (prostacyclin) and bosentan are used for pulmonary hypertension.
Calcium channel blockers, such as long-acting oral nifedipine (eg, 30 to 120 mg a day), may help Raynaud syndrome but may worsen gastric reflux. Bosentan, sildenafil, tadalafil, and vardenafil are other alternatives for severe Raynaud syndrome. Patients should dress warmly, wear mittens, and keep their head warm. IV infusions of prostaglandin E1 (alprostadil) or epoprostenol or sympathetic blockers can be used for severe digital ischemia. Treating superimposed infection is necessary to facilitate the healing of digital ulcers.
Renal manifestations
For acute renal crisis, a medical emergency, prompt treatment with an angiotensin-converting enzyme inhibitor can dramatically prolong survival (9). The mortality rate of scleroderma renal crisis remains high, but the crisis is often reversible if treatment is prompt.
Temporary or long-term dialysis may be necessary.
Renal transplantation is a feasible option in patients who develop end-stage renal disease.
Treatment references
1. Steen VD, Medsger TA Jr. Case-control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum. 1998;41(9):1613-1619. doi:10.1002/1529-0131(199809)41:9<1613::AID-ART11>3.0.CO;2-O
2. Jerjen R, Nikpour M, Krieg T, Denton CP, Saracino AM. Systemic sclerosis in adults. Part II: management and therapeutics. J Am Acad Dermatol. 2022;87(5):957-978. doi:10.1016/j.jaad.2021.10.066
3. van Laar JM, Farge D, Sont JK, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: A randomized clinical trial. JAMA. 2014;311(24):2490–2498. doi:10.1001/jama.2014.6368
4. Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): A randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4(9):708–719. doi:10.1016/S2213-2600(16)30152-7
5. Roofeh D, Lin CJF, Goldin J, et al. Tocilizumab prevents progression of early systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol. 2021;73(7):1301-1310. doi:10.1002/art.41668
6. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med. 2016;380(26):2518-2528. doi:10.1056/NEJMoa1903076
7. Maher TM, Tudor VA, Saunders P, et al. Rituximab versus intravenous cyclophosphamide in patients with connective tissue disease-associated interstitial lung disease in the UK (RECITAL): a double-blind, double-dummy, randomised, controlled, phase 2b trial. Lancet Respir Med. 2023;11(1):45-54. doi:10.1016/S2213-2600(22)00359-9
8. Pradère P, Tudorache I, Magnusson J, et al. Lung transplantation for scleroderma lung disease: An international, multicenter, observational cohort study. J Heart Lung Transplant. 2018;37(7):903-911. doi:10.1016/j.healun.2018.03.003
9. Steen VD, Costantino JP, Shapiro AP, Medsger TA Jr. Outcome of renal crisis in systemic sclerosis: relation to availability of angiotensin converting enzyme (ACE) inhibitors. Ann Intern Med. 1990;113(5):352-357. doi:10.7326/0003-4819-113-5-352
Prognosis for Systemic Sclerosis
In one cohort study, the overall 10-year survival was 92% for limited cutaneous systemic sclerosis and 65% for diffuse cutaneous systemic sclerosis (1). Predictors of early mortality include male sex, late onset, diffuse disease, pulmonary arterial hypertension, and renal crisis (2). The course depends on the type of disease (diffuse vs limited) and antibody profiling but can be unpredictable. Patients with diffuse cutaneous disease tend to have a more aggressive clinical course and eventually develop visceral complications (usually within the first 3 to 5 years), which, if severe, can lead to death. Heart failure may be intractable. Ventricular ectopy, even if asymptomatic, increases the risk of sudden death.
Patients with limited cutaneous systemic sclerosis (CREST syndrome—calcinosis cutis, Raynaud syndrome, esophageal dysmotility, sclerodactyly, telangiectasias) may have disease that is nonprogressive for long periods; visceral changes (eg, pulmonary hypertension caused by vascular disease of the lung, a peculiar form of biliary cirrhosis) eventually develop, but the course is often remarkably benign.
Prognosis references
1. Al-Dhaher FF, Pope JE, Ouimet JM. Determinants of morbidity and mortality of systemic sclerosis in Canada. Semin Arthritis Rheum. 2010;39(4):269-277. doi:10.1016/j.semarthrit.2008.06.002
2. Hao Y, Hudson M, Baron M, et al. Early mortality in a multinational systemic sclerosis inception cohort. Arthritis Rheumatol. 2017;69(5):1067–1077. doi:10.1002/art.40027
Key Points
Key findings in systemic sclerosis include skin and joint changes, Raynaud syndrome, and esophageal changes, but life-threatening manifestations may involve organs such as the lungs, heart, or kidneys.
Consider the diagnosis if patients have Raynaud syndrome, typical musculoskeletal or skin manifestations, or unexplained dysphagia, malabsorption, interstitial lung disease, pulmonary hypertension, cardiomyopathies, or conduction disturbances.
Test for ANA, Scl-70 (topoisomerase I), anticentromere, and anti-U3 RNP antibodies.
Because there is no clear disease-modifying therapy, direct treatment at involved organs.
Drug Information for the Topic





