



Cholestasis is absent or reduced bilirubin secretion, resulting in conjugated hyperbilirubinemia and jaundice. There are numerous causes, which are identified by laboratory testing, hepatobiliary scan, and, sometimes, liver biopsy and surgery. Treatment depends on cause.
Cholestasis occurs in 1/2500 neonates (1). It is defined as direct (conjugated) bilirubin > 1 mg/dL (> 17.1 micromol/L).
Cholestasis is never normal and warrants evaluation.
General reference
1. Amendola M, Squires JE. Pediatric Genetic Cholestatic Liver Disease Overview. 2022 Sep 15 [Updated 2023 May 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025.
Etiology of Neonatal Cholestasis
Cholestasis (see also Neonatal Hyperbilirubinemia) may result from extrahepatic or intrahepatic disorders, although some conditions overlap.
Extrahepatic causes of cholestasis
The most common extrahepatic disorder is:
Biliary atresia
Biliary atresia is obstruction of the biliary tree due to progressive sclerosis of the extrahepatic bile duct. In most cases, biliary atresia manifests several weeks after birth, probably after inflammation and scarring of the extrahepatic (and sometimes intrahepatic) bile ducts. It is rarely present in preterm infants or in neonates at birth.
Inspissated bile duct syndrome can also be a cause of extrahepatic neonatal cholestasis and is more common among infants with cystic fibrosis.
Biliary cysts rarely manifest as neonatal cholestasis; these cysts are more common among patients with autosomal recessive polycystic kidney disease (1).

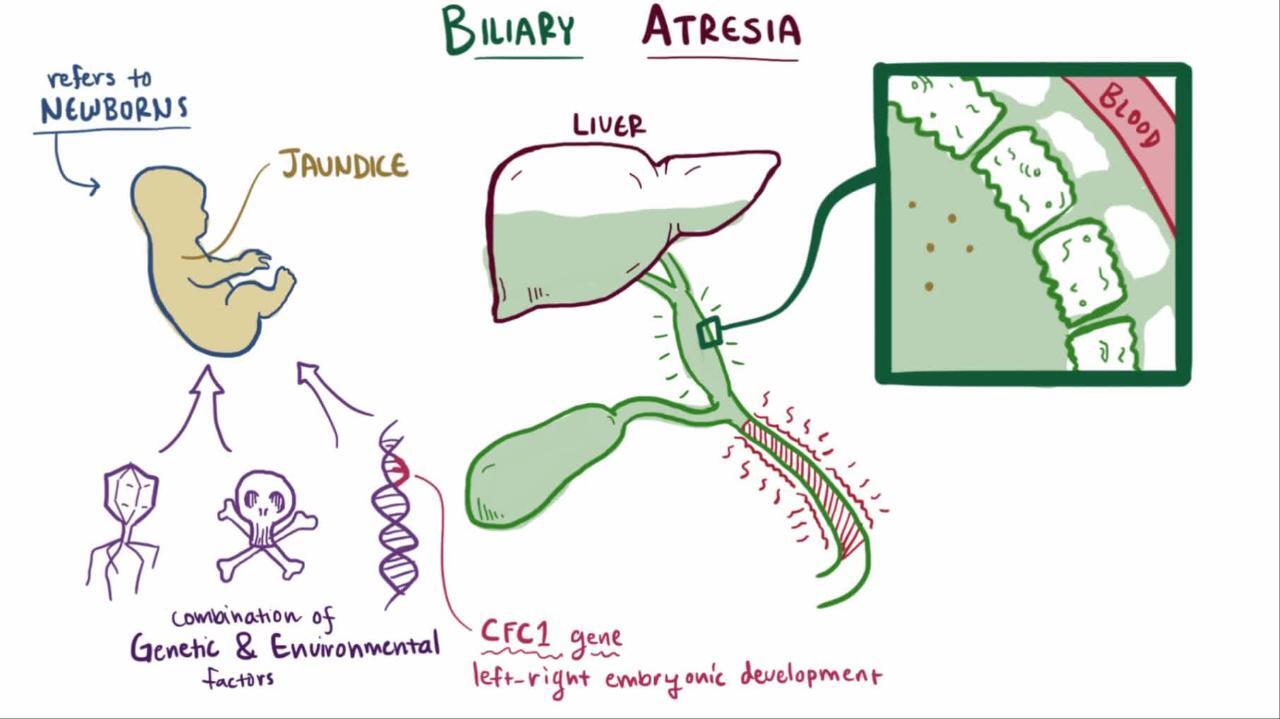
Intrahepatic causes of cholestasis
Intrahepatic causes can be infectious, alloimmune, metabolic/genetic, or toxic.
Infections can cause cholestasis. Infections may be viral (eg, herpes simplex virus, cytomegalovirus, rubella), bacterial (eg, gram-positive and gram-negative bacteremia, urinary tract infection caused by Escherichia coli), or parasitic (eg, toxoplasmosis). Sepsis in neonates receiving parental nutrition can also cause cholestasis.
Gestational alloimmune liver disease involves transplacental passage of maternal IgG that induces a complement-mediated membrane attack complex that injures the fetal liver.
Metabolic causes include numerous inborn errors of metabolism such as galactosemia, tyrosinemia, alpha-1 antitrypsin deficiency, disorders of lipid metabolism, bile acid defects, mitochondrial disorders, and fatty acid oxidation defects. Additional genetic defects include Alagille syndrome, cystic fibrosis, and arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. There are also a number of gene mutations that interfere with normal bile production and excretion and cause cholestasis; the resultant disorders are termed progressive familial intrahepatic cholestasis.
Toxic causes are due mainly to the use of prolonged parenteral nutrition in extremely preterm neonates or infants with short bowel syndrome. Newer generations of lipid emulsions in parenteral nutrition (eg, with fish oil and medium-chain triglycerides) appear to have decreased the risk of cholestasis (2).
Idiopathic neonatal hepatitis syndrome (giant cell hepatitis) is an inflammatory condition of the neonatal liver. Its incidence has decreased, and it is becoming rare because improved diagnostic studies allow identification of specific causes of cholestasis.
Etiology references
1. Fabris L, Fiorotto R, Spirli C, et al: Pathobiology of inherited biliary diseases: A roadmap to understand acquired liver diseases. Nat Rev Gastroenterol Hepatol 16(8):497–511, 2019. doi: 10.1038/s41575-019-0156-4
2. Guthrie G, Burrin D: Impact of Parenteral Lipid Emulsion Components on Cholestatic Liver Disease in Neonates. Nutrients 13(2):508, 2021. doi: 10.3390/nu13020508
Pathophysiology of Neonatal Cholestasis
In cholestasis, the primary failure is of bilirubin excretion, resulting in excess conjugated bilirubin in the bloodstream and decreased bile salts in the gastrointestinal (GI) tract. As a result of inadequate bile in the GI tract, there is malabsorption of fat and fat-soluble vitamins (A, D, E, and K), leading to vitamin deficiency, inadequate nutrition, and growth failure.
Symptoms and Signs of Neonatal Cholestasis
Cholestasis typically manifests in the first 2 weeks of life. Dark urine (containing conjugated bilirubin), acholic stools, and hepatomegaly can help distinguish cholestasis from physiologic jaundice and other causes of neonatal jaundice.
If cholestasis persists, chronic pruritus is common, as are symptoms and signs of fat-soluble vitamin deficiency; progression on growth charts may show a decline.
If the underlying disorder causes hepatic fibrosis and cirrhosis, portal hypertension with subsequent abdominal distention resulting from ascites, dilated abdominal veins, and upper GI bleeding resulting from esophageal varices may develop.
Diagnosis of Neonatal Cholestasis
Total and direct bilirubin
Liver tests
Tests for metabolic, infectious, and genetic causes
Liver ultrasound
Hepatobiliary scan
Occasionally biopsy of liver, operative cholangiography
Any infant who has jaundice after age 2 weeks (after age 3 weeks if breastfed and physical examination and urine and stool colors are normal) should be evaluated for cholestasis including with total and direct bilirubin levels (1). Some experts advocate that breastfed infants who have jaundice do not need to be evaluated until age 3 weeks. The initial approach should be directed at diagnosing treatable conditions (eg, extrahepatic biliary atresia, in which early surgical intervention improves short-term outcome).
Cholestasis is identified by an elevation in both total and direct bilirubin. Tests that are needed to further evaluate the liver include albumin, fractionated serum bilirubin, liver enzymes, prothrombin time/partial thromboplastin time (PT/PTT), and ammonia level (see Tests for Cholestasis).
Once cholestasis is confirmed, testing is required to determine etiology (see table ) and evidence of malabsorption (eg, low levels of the fat-soluble vitamins E, D, K, and A, or prolonged PT, suggesting a low level of vitamin K).
Diagnostic Evaluation for Neonatal Cholestasis
Etiology | Test |
|---|---|
Hepatic dysfunction | Albumin, ammonia, PT/PTT, AST, ALT, GGT, total and direct bilirubin (see Tests for Cholestasis) |
Infections | Urine cultures, TORCH titers, HIV screening and other hepatitides (hepatitis A, B, and C) |
Endocrinopathy | |
Sweat chloride test, review of newborn screening test | |
Neonatal screen, reducing substances (eg, galactose) in urine (see diagnosis of galactosemia) | |
Serum levels of alpha-1 antitrypsin, alpha-1 antitrypsin phenotype testing | |
Genetic errors in bile acid synthesis | Bile acid levels in urine and serum |
Urine organic acids, serum ammonia, serum electrolytes (see testing of inherited disorders of metabolism) | |
Alloimmune liver disease | Review of maternal obstetrical history for fetal deaths and/or prior infants with cholestasis Alpha fetoprotein, ferritin, lipid profile |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; GGT = gamma-glutamyl transpeptidase; prothrombin time/partial thromboplastin time = PT/PTT; TORCH = toxoplasmosis, other pathogens, rubella, cytomegalovirus, and herpes simplex; TSH = thyroid-stimulating hormone. | |
Abdominal ultrasound is often the first test; it is noninvasive and can assess liver size and certain abnormalities of the gallbladder and common bile duct. However, it is nonspecific.
A hepatobiliary scan using hydroxy iminodiacetic acid (HIDA scan; also called cholescintigraphy or hepatobiliary scintigraphy) should also be performed; excretion of contrast into the intestine rules out biliary atresia, but lack of excretion can occur with biliary atresia, severe neonatal hepatitis, and other causes of cholestasis. Infants with cholestasis are frequently given phenobarbital for 5 days prior to a HIDA scan in an attempt to enhance the excretion. In some tertiary pediatric centers, endoscopic retrograde cholangiopancreatography (ERCP) can be performed to investigate the anatomy of the biliary ductal system.
When no diagnosis has been made, a liver biopsy is generally performed relatively early on, sometimes with operative cholangiography. Patients with biliary atresia typically have enlarged portal triads, bile duct proliferation, and increased fibrosis. Neonatal hepatitis is characterized by lobular disarray with multinucleated giant cells. Alloimmune liver disease is characterized by elevated hepatic iron stores.
Diagnosis reference
1. Fawaz R, Baumann U, Ekong U, et al. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2017;64(1):154-168. doi:10.1097/MPG.0000000000001334
Treatment of Neonatal Cholestasis
Specific cause treated
Vitamin A, D, E, and K supplements
Medium-chain triglycerides
Sometimes ursodeoxycholic acid
For suspected biliary atresia, surgical exploration and sometimes portoenterostomy (Kasai procedure)
Specific treatment is directed at the cause. If there is no specific therapy, treatment is supportive and consists primarily of nutritional therapy, including supplements of vitamins A, D, E, and K (1).
For formula-fed infants, a formula that is high in medium-chain triglycerides should be used because it is absorbed better in the presence of bile salt deficiency. Adequate calories are required; infants may need > 130 calories/kg/day. In infants with some bile flow, ursodeoxycholic acid once or twice a day may relieve itching, improve bile flow, and potentially improve liver disease.
Infants with presumed biliary atresia require surgical exploration with an intraoperative cholangiogram (2). If biliary atresia is confirmed, a portoenterostomy (Kasai procedure) should be performed. Ideally, this procedure should be performed in the first month of life. After this period, the prognosis significantly worsens. Postoperatively, many patients have significant chronic problems, including persistent cholestasis, recurrent ascending cholangitis, and growth and weight faltering (formerly failure to thrive). Prophylactic antibiotics (eg, trimethoprim/sulfamethoxazole) are frequently prescribed for a year postoperatively in an attempt to prevent ascending cholangitis. Even with optimal therapy, many patients develop cirrhosis and ultimately require liver transplantation.
Cholestasis caused by parenteral nutrition resolves if the parenteral nutrition is stopped or sometimes if a newer-generation lipid emulsion is substituted for an older one before the infant develops severe liver disease.
Because gestational alloimmune liver disease has no definitive marker or test, treatment with IV immune globulin (IVIG) or exchange transfusion needs to be considered early to reverse the ongoing liver injury if no definite diagnosis has been made (3).
Treatment references
1. Mouzaki M, Bronsky J, Gupte G, et al. Nutrition Support of Children With Chronic Liver Diseases: A Joint Position Paper of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2019;69(4):498-511. doi:10.1097/MPG.0000000000002443
2. Tam PKH, Wells RG, Tang CSM, et al. Biliary atresia. Nat Rev Dis Primers. 2024;10(1):47. Published 2024 Jul 11. doi:10.1038/s41572-024-00533-x
3. Fischer HS, Staufner C, Sallmon H, et al. Early exchange transfusion to treat neonates with gestational alloimmune liver disease: An 11-year cohort study. J Pediatr Gastroenterol Nutr. 2020;70(4):444–449. doi:10.1097/MPG.0000000000002593
Prognosis for Neonatal Cholestasis
Prognosis for cholestasis due to specific disorders (eg, metabolic disease) is variable, ranging from a completely benign course to a progressive disease resulting in cirrhosis.
Biliary atresia is progressive and, if untreated, results in cirrhosis with portal hypertension by several months of age, then liver failure and death by 1 year of age.
Idiopathic neonatal hepatitis syndrome usually resolves slowly, but permanent liver damage may result and lead to liver failure and death.
Gestational alloimmune liver disease has a poor prognosis without early intervention.
Key Points
There are numerous inherited and acquired causes of neonatal cholestasis, resulting in failure of bilirubin excretion and thus excess conjugated bilirubin.
Neonatal cholestasis typically is noted in the first 2 weeks of life; infants have jaundice and often have dark urine, acholic stools, and hepatomegaly.
Begin with laboratory testing of the liver, ultrasound, hepatobiliary scan, and tests for specific causes, sometimes including liver biopsy.
Treat specific cause and give supportive care, including supplementation of fat-soluble vitamins and, for formula-fed infants, a formula that is high in medium-chain triglycerides and contains sufficient calories.
Drug Information for the Topic





