



Cystic fibrosis is an inherited disease of the exocrine glands affecting primarily the gastrointestinal and respiratory systems. It leads to chronic lung disease, exocrine pancreatic insufficiency, hepatobiliary disease, and abnormally high sweat electrolytes. Diagnosis is by sweat test or identification of 2 cystic fibrosis–causing gene variants in patients with a positive newborn screening test result or characteristic clinical features. Treatment is supportive through aggressive multidisciplinary care along with small-molecule correctors and potentiators targeting the cystic fibrosis transmembrane conductance regulator protein defect.
Cystic fibrosis (CF) is a life-threatening genetic disease, which in the United States occurs in approximately 1/3,300 births of White infants, 1/15,300 births of Black infants, and 1/32,000 births of Asian American infants as of 2021(1). There are approximately 40,000 people living with CF in the United States and an estimated 105,000 people have been diagnosed with CF across 94 countries worldwide (2). Because of improved treatment and life expectancy, approximately 60% of patients in the United States with CF are adults.
General references
1. Cystic Fibrosis Foundation Patient Registry 2021 Annual Data Report Bethesda, Maryland 2025 Cystic Fibrosis Foundation. Accessed July 30, 2025.
2 Cystic Fibrosis Foundation: About Cystic Fibrosis. Accessed July 30, 2025.
Etiology of Cystic Fibrosis
Cystic fibrosis is inherited as an autosomal recessive trait by approximately 4% (1 in 27) of the White population (1). The gene responsible for CF has been localized on the long arm of chromosome 7. It encodes a membrane-associated protein called the cystic fibrosis transmembrane conductance regulator (CFTR). The most common gene variant, F508del, occurs in about 85% of CF alleles; > 2000 less common CFTR variants have been identified (2).
CFTR is a cyclic adenosine monophosphate (cAMP)–regulated chloride channel, regulating chloride, sodium, and bicarbonate transport across epithelial membranes. It is expressed in multiple organ systems, including the respiratory tract, pancreas, intestine, and sweat glands (see Pathophysiology of Cystic Fibrosis). A number of additional functions are considered likely. Disease manifests only in people who are homozygous (ie, biallelic pathogenic variants). People who are heterozygous may show subtle abnormalities of epithelial electrolyte transport but are clinically unaffected.
CFTR variants can involve frameshift (a deletion or insertion in a DNA sequence that shifts the way a sequence is read) or nonsense (stop) mutations. These variants have been divided into 6 classes based on how the variant affects the function or processing of the CFTR protein (3). Patients with class I, II, or III variants are considered to have a more severe genotype that results in little or no CFTR function, whereas patients with 1 or 2 class IV, V, or VI variants are considered to have a milder genotype that results in residual CFTR function. However, there is no strict relationship between specific variants and disease manifestation, so clinical testing (ie, of organ function) rather than genotyping is considered a better guide to prognosis.
Etiology references
1. Zvereff VV, Faruki H, Edwards M, Friedman KJ: Cystic fibrosis carrier screening in a North American population. Genet Med 16(7):539-546, 2014. doi: 10.1038/gim.2013.188
2. Grasemann H, Ratjen F: Cystic Fibrosis. N Engl J Med 389(18):1693-1707, 2023. doi:10.1056/NEJMra2216474
3. Ong T, Ramsey BW: Cystic Fibrosis: A Review. JAMA 329(21):1859-1871, 2023. doi:10.1001/jama.2023.8120
Pathophysiology of Cystic Fibrosis
Nearly all exocrine glands are affected in varying distribution and degree of severity. Glands may:
Become obstructed by viscid mucus in the lumen (pancreas, intestinal glands, intrahepatic bile ducts, gallbladder, and submaxillary glands)
Appear histologically abnormal and produce excessive secretions (tracheobronchial and Brunner glands)
Appear histologically normal but secrete excessive sodium and chloride (sweat, parotid, and small salivary glands)


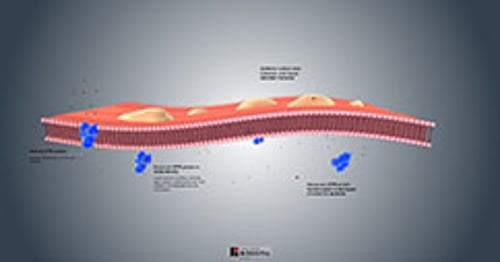
Section A of the illustration shows the organs that may be affected by cystic fibrosis.
Section B shows a normal airway with a thin layer of mucus lining the airway epithelial wall.
Section C shows an airway with cystic fibrosis. The widened airway is blocked by thick, viscid mucus that contains sanguineous or purulent material, often including infectious organisms such as bacteria.
Source: National Heart, Lung, and Blood Institute; National Institutes of Health; U.S. Department of Health and Human Services.
Respiratory
Although the lungs are generally histologically normal at birth, most patients develop signs or symptoms of pulmonary disease beginning in infancy or early childhood. Mucus plugging and chronic bacterial infection, accompanied by a pronounced inflammatory response, damage the airways, ultimately leading to bronchiectasis and respiratory insufficiency. The clinical course of the disease is characterized by episodic exacerbations with infection and progressive decline in pulmonary function.
Pulmonary damage is probably initiated by diffuse obstruction in the small airways by abnormally thick mucus secretions. Bronchiolitis and mucopurulent plugging of the airways occur secondary to obstruction and infection. Chronic inflammation secondary to the release of proteases and proinflammatory cytokines by cells in the airways also contributes to lung injury. Airway changes are more common than parenchymal changes, and emphysema is not prominent. About 40% of patients have bronchial hyperreactivity that may partially or not at all respond to bronchodilators (1, 2).
In patients with advanced pulmonary disease, chronic hypoxemia results in muscular hypertrophy of the pulmonary arteries, pulmonary hypertension, and right ventricular hypertrophy.
The lungs of most patients are colonized by pathogenic bacteria. Early in the course, Staphylococcus aureus is the most common pathogen, but as the disease progresses, Pseudomonas aeruginosa, including multidrug-resistant strains, is frequently isolated. A mucoid variant of P. aeruginosa is uniquely associated with CF and results in a worse prognosis than nonmucoid P. aeruginosa.
In the United States, the prevalence of methicillin-resistant S. aureus (MRSA) in the respiratory tract is approximately 25% (3). Patients chronically infected with MRSA typically have more rapid declines in pulmonary function and lower survival rates than those who are not .
In the United States, colonization with Burkholderia cepacia complex occurs in about 1.2% of patients as of 2023 and may be associated with more rapid pulmonary deterioration (4).
Nontuberculous mycobacteria, including Mycobacterium avium complex and M. abscessus, are potential respiratory pathogens. Differentiating infection from colonization can be challenging.
Other common respiratory pathogens include Stenotrophomonas maltophilia, Achromobacter xylosoxidans, and Aspergillus species.
Anaerobic bacteria and common respiratory viruses are frequently present in the respiratory tract of patients with CF, but their role in disease progression has not been well established.
Gastrointestinal
The pancreas, intestines, and hepatobiliary system are frequently affected. Exocrine pancreatic function is compromised in approximately 85% of patients (5). An exception is a subset of patients who have certain CFTR variants with residual function (ie, class IV, V, or VI variants), in whom pancreatic function is preserved. Patients with pancreatic insufficiency have malabsorption of fats, fat-soluble vitamins, and protein which leads to steatorrhea, malnutrition, and growth faltering in children. Duodenal fluid is abnormally viscid and shows an absence or diminution of enzyme activity and decreased bicarbonate concentration; stool trypsin and chymotrypsin are also absent or diminished. Endocrine pancreatic dysfunction is less common, but impaired glucose tolerance or diabetes mellitus is present in approximately 20% of adolescents and up to 50% of adults (6).
Cystic fibrosis related liver disease (including hepatic steatosis) occurs in approximately 30% of patients (7). Bile duct involvement with bile stasis and biliary plugging leads to hepatic fibrosis. Approximately 3 to 4% of patients progress to irreversible multinodular cirrhosis with varices and portal hypertension, usually by 12 years of age. Hepatocellular failure is a rare and late event. There is an increased incidence of cholelithiasis, which is usually asymptomatic.
Abnormally viscid intestinal secretions can cause meconium ileus in neonates and sometimes meconium plugging of the colon. Older children and adults also may have intermittent or chronic constipation and intestinal obstruction.
Other gastrointestinal (GI) problems include intussusception, volvulus, rectal prolapse, periappendiceal abscess, pancreatitis, an increased risk of cancer of the hepatobiliary tract and cancer of the GI tract (including of the pancreas), gastroesophageal reflux, esophagitis, and an increased prevalence of Crohn disease and celiac disease.
Other
Infertility occurs in approximately 98% of adult males secondary to maldevelopment or congenital absence of the vas deferens or to other forms of obstructive azoospermia (8). However, sperm production is typically preserved. In women, fertility is somewhat decreased secondary to viscid cervical secretions, although many women have carried pregnancies to term. Pregnancy outcome for both the mother and neonate is related to the mother's health.
Other complications include chronic rhinosinusitis, osteopenia/osteoporosis, depression and anxiety, chronic pain, obstructive sleep apnea, other sleep disorders, renal stones, dialysis-dependent chronic kidney disease (possibly related to treatments as well as to CF), iron deficiency anemia, sensorineural hearing loss and tinnitus caused by exposure to ototoxic medications (especially aminoglycosides), and episodic arthralgias/arthritis.
Pathophysiology references
1. Levine H, Cohen-Cymberknoh M, Klein N, et al: Reversible airway obstruction in cystic fibrosis: Common, but not associated with characteristics of asthma. J Cyst Fibros 15(5):652-659, 2016. doi:10.1016/j.jcf.2016.01.003
2. McCuaig S, Martin JG: How the airway smooth muscle in cystic fibrosis reacts in proinflammatory conditions: implications for airway hyper-responsiveness and asthma in cystic fibrosis. Lancet Respir Med 1(2):137-147, 2013. doi:10.1016/S2213-2600(12)70058-9
3. Chmiel JF, Aksamit TR, Chotirmall SH, et al: Antibiotic management of lung infections in cystic fibrosis. I. The microbiome, methicillin-resistant Staphylococcus aureus, gram-negative bacteria, and multiple infections. Ann Am Thorac Soc 11(7):1120-1129, 2014. doi:10.1513/AnnalsATS.201402-050AS
4. Cystic Fibrosis Foundation: Patient Registry: 2023 Patient Registry and Annual Data Report. September 2024. Accessed August 7, 2025.
5. Ritivoiu ME, Drăgoi CM, Matei D, et al: Current and Future Therapeutic Approaches of Exocrine Pancreatic Insufficiency in Children with Cystic Fibrosis in the Era of Personalized Medicine. Pharmaceutics 15(1):162, 2023. doi:10.3390/pharmaceutics15010162
6. Mason KA, Marks BE, Wood CL, Le TN: Cystic fibrosis-related diabetes: The patient perspective. J Clin Transl Endocrinol 26:100279, 2021. doi:10.1016/j.jcte.2021.100279
7. Coman DE, Vincent C, Lavoie A, Bilodeau M, Hercun J: A82 PREVALENCE AND NON-INVASIVE SCREENING FOR CYSTIC FIBROSIS RELATED LIVER DISEASE IN A COHORT FOLLOWED AT A CYSTIC FIBROSIS REFERENCE CENTER. J Can Assoc Gastroenterol 6(Suppl 1):44-45, 2023. doi:10.1093/jcag/gwac036.082
8. Cystic Fibrosis Foundation: Fertility in Men with CF. Accessed July 30, 2025.
Symptoms and Signs of Cystic Fibrosis
Respiratory
Recurrent or chronic infections presenting with cough, sputum production, and wheezing are common. Cough is the most common chronic symptom, often accompanied by sputum production.. Intercostal retractions, the use of accessory muscles of respiration, a barrel-chest deformity, digital clubbing, cyanosis, and a declining tolerance for exercise occur with disease progression. Upper respiratory tract involvement may include nasal polyposis and chronic rhinosinusitis or recurrent episodes of acute sinusitis.
Pulmonary complications include pneumothorax, nontuberculous mycobacterial infection, hemoptysis, allergic bronchopulmonary aspergillosis (ABPA), and cor pulmonale (right heart failure secondary to pulmonary hypertension).
Gastrointestinal
Meconium ileus due to obstruction of the ileum by viscid meconium may be the earliest sign and is present in approximately 20% of CF-affected neonates (1). It typically manifests with abdominal distention, vomiting, and failure to pass meconium. Some infants may also develop intestinal perforation, with signs of peritonitis and shock. Infants with meconium plug syndrome have a delayed passage of meconium. They can have similar signs of obstruction or very mild and transient symptoms that go unnoticed. In infants without meconium ileus, disease onset may be heralded by a delay in regaining birth weight and inadequate weight gain at 4 to 6 weeks of age.
Occasionally, infants who are undernourished, especially if on hypoallergenic formula or soy formula, present with generalized edema secondary to protein malabsorption.
Pancreatic insufficiency is usually clinically apparent early in life and may be progressive. Manifestations include the frequent passage of steatorrheic stools (bulky, foul-smelling, oily stools); abdominal protuberance; and poor growth pattern with decreased subcutaneous tissue and muscle mass despite a normal or voracious appetite. Clinical manifestations may occur secondary to deficiency of fat-soluble vitamins A, D, E and K. For further information, see Overview of Vitamins.
Rectal prolapse may occur in untreated infants and toddlers due to the combination of increased intra-abdominal pressure secondary to coughing, and strain involved in passing bulky stools.
Gastroesophageal reflux is relatively common among children and adults.
Constipation is another common issue among patients with CF. In addition, patients of all ages are at risk of developing distal intestinal obstruction syndrome, which is characterized by recurrent and sometimes chronic episodes of partial or complete small- or large-bowel obstruction. Both constipation and distal intestinal obstruction syndrome likely occur due to an accumulation of inspissated mucoid secretions in the bowel. Symptoms include cramps, abdominal pain, change in stooling pattern, decreased appetite, and sometimes vomiting.
Other
Excessive sweating in hot weather or with fever may lead to episodes of hyponatremic/hypochloremic dehydration and circulatory failure. In arid climates, infants may present with chronic metabolic alkalosis. Salt crystal formation and a salty taste on the skin are highly suggestive of CF.
Children may have disturbed sleep due to poor sleep quality and efficiency (2). Causes of disturbed sleep may include nocturnal cough, chronic pain, sleep disordered breathing, and sleep apnea.
Adolescents may have retarded growth and delayed onset of puberty.
Symptoms and signs references
1. Sathe M, Houwen R: Meconium ileus in Cystic Fibrosis. J Cyst Fibros 16 Suppl 2:S32-S39, 2017. doi:10.1016/j.jcf.2017.06.007
2. Reiter J, Breuer O, Cohen-Cymberknoh M, Forno E, Gileles-Hillel A: Sleep in children with cystic fibrosis: More under the covers. Pediatr Pulmonol 57(8):1944-1951, 2022. doi:10.1002/ppul.25462
Diagnosis of Cystic Fibrosis
Newborn screening
May also be suggested by a positive prenatal screening test result, family history, or symptomatic presentation
Confirmed by a sweat test showing elevated sweat chloride on 2 occasions
Identifying 2 CF-causing variants (1 on each chromosome) is consistent with the diagnosis
May rarely be confirmed, in atypical cases, by demonstrating abnormal ion transport across the nasal epithelium or abnormal intestinal current measurements
Most cases of cystic fibrosis are first identified by newborn screening where such testing is available and well utilized. In regions where newborn screening is not widely available, patients may sometimes not be officially diagnosed with CF until adolescence or early adulthood. Despite advances in genetic testing, the sweat chloride test remains the standard for confirming a CF diagnosis in most cases because of its sensitivity and specificity, simplicity, and availability (1).
Newborn screening
Universal newborn screening for CF is standard in the United States (2). In the United States, mandatory newborn screening for cystic fibrosis has accounted for an increase in detection of cystic fibrosis to 54.1% of all new diagnoses and 85.1% of diagnoses among those less than 6 months old (3). Screening is based on detecting an elevated concentration of immunoreactive trypsinogen (IRT) in the blood. Newborn screening in the United States has been associated with improved outcomes including improved nutritional status up to age 10 years, a faster increase in lung function, and older age at chronic P. aeruginosa infection (4).
There are 2 methods of following up on an elevated IRT level. In one method, a second IRT is done, which, if also elevated, is followed by a DNA testing for the CFTR mutation (also called the IRT-IRT-DNA method), followed by a sweat chloride test (2). In the other, more commonly used method, one elevated IRT level is followed directly by DNA CFTR mutation testing, and, if 1 or 2 variants are identified, then a sweat chloride test is done (also called the IRT-DNA method). The IRT-IRT-DNA method has greater sensitivity than the IRT-DNA method (5).
Neonates with a positive newborn screen or prenatal genetic test should ideally undergo sweat chloride testing by the age of 4 weeks to ensure that any related disease manifestations are detected early and treated quickly (6).
Sweat chloride testing
To perform the test, localized sweating is stimulated with pilocarpine iontophoresis, and the chloride concentration in the collected sweat is measured. Although the sweat chloride concentration increases slightly with age, the sweat test may be performed at almost all ages:
Normal: ≤ 30 mEq/L (≤ 30 mmol/L) (CF is unlikely.)
Intermediate: 30 to 59 mEq/L (30 to 59 mmol/L) (CF is possible.)
Abnormal: ≥ 60 mEq/L (≥ 60 mmol/L) (This result is consistent with CF.)
The test should not be performed before 48 hours of life and is ideally done between 2 and 4 weeks of age. An adequate sweat sample (> 75 mg on filter paper or > 15 mcL in microbore tubing) is difficult to obtain before 2 weeks of age. False-negative results are rare but may occur in the presence of edema and hypoproteinemia or an inadequate quantity of sweat. False-positive results are usually due to technical error. Transient elevation of sweat chloride concentration can result from psychosocial deprivation (eg, child abuse, neglect) and can occur in patients with anorexia nervosa. A positive sweat test result should be confirmed by a second sweat test or by identification of 2 CF-causing gene mutations.
Intermediate sweat test results
A small subset of patients have a mild or partial CF phenotype and sweat chloride values that are persistently in the intermediate or even normal range. In addition, there are patients who have single-organ manifestations such as chronic or recurrent pancreatitis, isolated bronchiectasis, or congenital bilateral absence of the vas deferens along with findings suggestive of abnormal CFTR function. Such patients do not meet criteria for a CF diagnosis and are classified as having a CFTR-related disorder. In some, the diagnosis of CF can be confirmed by the identification of 2 CF-causing gene mutations (variants), 1 on each chromosome. If 2 CF-causing variants are not identified, ancillary evaluations such as pancreatic function testing and pancreatic imaging, high-resolution chest CT, sinus CT, pulmonary function testing, urogenital evaluation in males, and bronchoalveolar lavage including assessment of microbial flora may be useful to complement the diagnosis.
Additional potentially helpful diagnostic tests include expanded CFTR genetic analysis and measurement of nasal transepithelial potential difference (based on the observation of increased sodium reabsorption across epithelium that is relatively impermeable to chloride in patients with CF) and measurement of intestinal currents.
CFTR-related metabolic syndrome and CF screen positive, inconclusive diagnosis
Infants who have a positive newborn screening result and evidence of possible CFTR dysfunction but do not meet the diagnostic criteria for CF are classified as having CFTR-related metabolic syndrome (CRMS), also called CF screen positive, inconclusive diagnosis (CFSPID). CRMS/CFSPID is diagnosed in infants who have a positive newborn screen, are asymptomatic, and have either of the following (7):
Sweat chloride concentrations in the intermediate range and 0 or 1 CF-causing variant
Sweat chloride concentrations in the normal range and 2 CFTR variants, at least 1 of which has unclear phenotypic consequences
Most children with CRMS/CFSPID remain healthy, but over time < 10% may develop symptoms (eg, pulmonary disease) and meet criteria for a diagnosis of CF or a CF-related disorder (1). Patients with CRMS/CFSPID should be evaluated and monitored regularly in a CF care center.
Pancreatic tests
Pancreatic function should be assessed at the time of diagnosis, usually by measuring the concentration of human pancreatic elastase in stool. Human pancreatic elastase measurement may be performed even in the presence of exogenous pancreatic enzymes. Infants who are initially pancreatic sufficient and who carry 2 "severe" variants should have serial measurements of pancreatic enzymes approximately every 6 to 12 months to detect progression to pancreatic insufficiency (8).
Respiratory assessment
Chest imaging is performed at times of pulmonary deterioration or exacerbations and routinely every 1 to 2 years. High-resolution chest CT may be helpful to more precisely define the extent of lung damage and to detect subtle airway abnormalities. Chest radiographs and CT may show hyperinflation, mucoid impaction, and bronchial wall thickening as the earliest findings. Subsequent changes include areas of infiltrate, atelectasis, and hilar adenopathy. With advanced disease, segmental or lobar atelectasis, cyst formation, bronchiectasis, and pulmonary artery and right ventricular hypertrophy occur. Branching, fingerlike opacifications that represent mucoid impaction of dilated bronchi are characteristic.
Sinus CT studies are indicated in patients with significant sinus symptoms or nasal polyps in whom endoscopic sinus surgery is being considered. These studies almost always show persistent opacification of the paranasal sinuses, suggesting chronic rhinosinusitis.

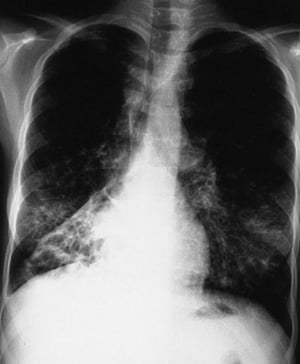
This chest radiograph shows right lower lobe collapse. Findings are typical for CF but not specific.
This chest radiograph shows right lower lobe collapse. Findings are typical for CF but not specific.
By permission of the publisher. From Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.

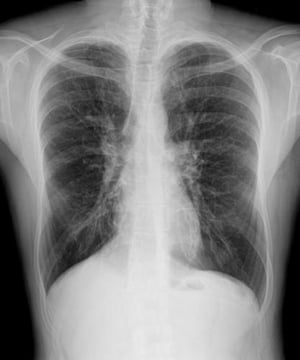
This chest radiograph of a man with cystic fibrosis shows increased lung markings consistent with bronchiectasis.
This chest radiograph of a man with cystic fibrosis shows increased lung markings consistent with bronchiectasis.
PHOTOSTOCK-ISRAEL/SCIENCE PHOTO LIBRARY

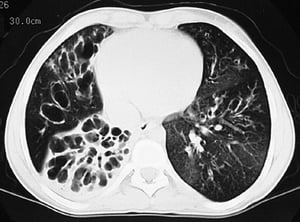
This CT scan shows greatly dilated bronchi throughout the lungs. Findings are typical for CF but not specific.
This CT scan shows greatly dilated bronchi throughout the lungs. Findings are typical for CF but not specific.
By permission of the publisher. From Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.
This chest radiograph shows right lower lobe collapse. Findings are typical for CF but not specific.
This chest radiograph shows right lower lobe collapse. Findings are typical for CF but not specific.
By permission of the publisher. From Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.
This chest radiograph of a man with cystic fibrosis shows increased lung markings consistent with bronchiectasis.
This chest radiograph of a man with cystic fibrosis shows increased lung markings consistent with bronchiectasis.
PHOTOSTOCK-ISRAEL/SCIENCE PHOTO LIBRARY
This CT scan shows greatly dilated bronchi throughout the lungs. Findings are typical for CF but not specific.
This CT scan shows greatly dilated bronchi throughout the lungs. Findings are typical for CF but not specific.
By permission of the publisher. From Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.
Pulmonary function tests are the best indicators of clinical status and response to therapy. In patients over 5 years of age, spirometry should be done routinely and whenever clinical decline is suspected. In infants, respiratory status can be monitored by using a raised-volume rapid thoracoabdominal compression technique, which generates a partial flow-volume curve. In children 3 to 6 years of age, the multiple breath washout procedure can be used to generate a lung clearance index as a measure of ventilation inhomogeneity (9).
Pulmonary function tests done by spirometry indicate:
A reduction in forced vital capacity (FVC), forced expiratory volume in 1 second (FEV1), forced expiratory flow between 25% and 75% expired volume (FEF25-75), and FEV1/FVC ratio
An increase in residual volume and the ratio of residual volume to total lung capacity
Screening oropharyngeal or sputum cultures should be done at least 4 times/year, especially in patients not yet colonized with P. aeruginosa. Bronchoscopy/bronchoalveolar lavage is indicated when it is important to precisely define the patient’s lower airway microbial flora (eg, to direct antibiotic selection) or to remove inspissated mucus plugs.
Carrier screening
CF carrier screening is available in the United States and is recommended for couples who are planning a pregnancy or seeking prenatal care. If both potential parents carry a CFTR variant, prenatal screening of the fetus can be done by chorionic villus sampling or amniocentesis. Prenatal counseling in such cases is complicated by the wide phenotypic variability of CF and incomplete information on the clinical consequences of many of the CFTR variants that are identified through screening. Confirmatory testing with sweat chloride analysis is usually performed in infants with a positive or inconclusive maternofetal carrier screen to establish the diagnosis.
Diagnosis references
1. Green DM, Lahiri T, Raraigh KS, et al: Cystic Fibrosis Foundation Evidence-Based Guideline for the Management of CRMS/CFSPID. Pediatrics 153(5):e2023064657, 2024. doi:10.1542/peds.2023-064657
2. McGarry ME, Raraigh KS, Farrell P, et al: Cystic Fibrosis Newborn Screening: A Systematic Review-Driven Consensus Guideline from the United States Cystic Fibrosis Foundation. Int J Neonatal Screen 11(2):24, 2025. doi:10.3390/ijns11020024
3. Cystic Fibrosis Foundation: Patient Registry. 2023 Annual Data Report Bethesda, Maryland. September 2024. Accessed August 7, 2025
4. Rosenfeld M, Ostrenga J, Cromwell EA, et al: Real-world Associations of US Cystic Fibrosis Newborn Screening Programs With Nutritional and Pulmonary Outcomes. JAMA Pediatr 176(10):990-999, 2022. doi:10.1001/jamapediatrics.2022.2674
5. Sontag MK, Lee R, Wright D, Freedenberg D, Sagel SD: Improving the Sensitivity and Positive Predictive Value in a Cystic Fibrosis Newborn Screening Program Using a Repeat Immunoreactive Trypsinogen and Genetic Analysis. J Pediatr 175:150-158.e1, 2016. doi:10.1016/j.jpeds.2016.03.046
6. Cystic Fibrosis Foundation: Newborn Screening for CF. Accessed July 30, 2025.
7. Cystic Fibrosis Foundation: CFTR-Related Metabolic Syndrome (CRMS). Accessed July 30, 2025.
8. Abu-El-Haija M, Uc A, Werlin SL, et al: Nutritional Considerations in Pediatric Pancreatitis: A Position Paper from the NASPGHAN Pancreas Committee and ESPGHAN Cystic Fibrosis/Pancreas Working Group. J Pediatr Gastroenterol Nutr 67(1):131-143, 2018. doi:10.1097/MPG.0000000000002023
9. Stanojevic S, Davis SD, Retsch-Bogart G, et al: Progression of lung disease in preschool patients with cystic fibrosis. Am J Respir Crit Care Med 195:1216–1225, 2017. doi: 10.1164/rccm.201610-2158OC
Treatment of Cystic Fibrosis
Comprehensive, multidisciplinary support
Antibiotics, inhaled medications to thin airway secretions, and physical maneuvers to clear airway secretions
Inhaled bronchodilators and sometimes corticosteroids (glucocorticoids) for responders
Usually pancreatic enzyme and vitamin supplementation
High-calorie diet (sometimes requiring supplemental enteral tube feedings)
In patients with specific variants, CFTR modulators consisting of a CFTR potentiator or combination of CFTR correctors and a CFTR potentiator
Adjunctive measures (eg, surgery, liver transplant)
Comprehensive and intensive therapy should be directed by an experienced physician working with a multidisciplinary team that includes other physicians, nurses, dietitians, physical and respiratory therapists, mental health professionals, pharmacists, and social workers. The goals of therapy are maintenance of normal nutritional status, prevention or aggressive treatment of pulmonary and other complications, encouragement of physical activity, and provision of psychosocial support. The treatment regimen is complex and may take up to 2 hours each day. With appropriate support, most patients can make an age-appropriate adjustment at home and school.
Treatment of respiratory manifestations
Treatment of pulmonary manifestations should focus on prevention of airway obstruction and prophylaxis and control of pulmonary infections. In the United States, prophylaxis against pulmonary infections involves maintenance of pertussis, Haemophilus influenzae, varicella, Streptococcus pneumoniae, and measles immunity; annual influenza vaccination; and appropriate COVID-19 vaccination.
In patients exposed to influenza, a neuraminidase inhibitor (eg, oseltamivir) can be used prophylactically or at the first signs of infection. Giving nirsevimab, or when not available, palivizumab, to infants with CF may provide additional protection against respiratory syncytial virus infection. Clinical data on RSV prevention medications in patients with CF is limited; therefore, careful clinical judgment is warranted before their administration.
Long-term daily inhalation therapy with dornase alfa (recombinant human deoxyribonuclease) or with 7% hypertonic saline is recommended (1, 2); it slows the rate of decline in pulmonary function and decreases the frequency of respiratory tract exacerbations (3).
Airway clearance measures consisting of postural drainage, percussion, vibration, and assisted coughing (chest physiotherapy) are recommended at the time of diagnosis and should be done on a regular basis. In older patients, alternative airway clearance measures, such as active cycles of breathing, autogenic (self-managed) drainage, positive expiratory pressure devices, and vest therapy (involving high-frequency chest wall oscillation), may be effective. Regular aerobic exercise is recommended when possible to help airway clearance. For patients with obstructive sleep apnea, continuous positive airway pressure may be beneficial.
For patients with reversible airway obstruction, bronchodilators may be given by inhalation. Corticosteroids (glucocorticoids) by inhalation are usually not effective. Oxygen therapy is indicated for patients with severe pulmonary insufficiency and hypoxemia.
Oral expectorants are sometimes used, but limited data support their efficacy. Cough suppressants should be discouraged.
Oral glucocorticoids are indicated in infants with prolonged bronchiolitis and in patients with refractory bronchospasm, allergic bronchopulmonary aspergillosis (ABPA), or inflammatory complications (eg, arthritis, vasculitis). Long-term use of alternate-day glucocorticoid therapy can slow the decline in pulmonary function, but because of glucocorticoid-related complications, it is not recommended for routine use. Patients receiving glucocorticoids must be closely monitored for evidence of diabetes and linear stunting of growth. Allergic bronchopulmonary aspergillosis is additionally treated with an oral or IV antifungal medication; duration of treatment may sometimes be prolonged.
Ibuprofen, when given over several years at a dose sufficient to achieve a peak plasma concentration between 50 and 100 mcg/mL (242.4 and 484.8 micromol/L), has been shown to slow the rate of decline in pulmonary function, especially in children 5 to 13 years of age. The appropriate dose must be individualized based on pharmacokinetic studies.
Chronic rhinosinusitis may be treated with nasal saline irrigation, low-pressure isotonic nasal irrigation, intranasal dornase alfa nebulization, and/or sinonasal topical antibiotics. Sinus surgery may be helpful in cases refractory to medical management. An intranasal glucocorticoid spray is recommended to treat allergic rhinitis.
Noninvasive positive pressure ventilation nasally or by face mask can be beneficial in appropriately selected patients with acute respiratory failure. Mechanical ventilation or extracorporeal membrane oxygenation (ECMO) is typically not indicated for end-stage chronic respiratory failure unless it is used as a bridge to lung transplantation or in patients with good or moderate baseline status in whom acute reversible respiratory complications develop.
Pneumothorax can be treated with closed chest tube thoracostomy drainage. Open thoracotomy or thoracoscopy with resection of pleural blebs and mechanical abrasion of the pleural surfaces is effective in treating recurrent pneumothoraces.
Mild to moderate hemoptysis is treated with antibiotics (oral/aerosol or IV depending on severity of hemoptysis and severity of infection) and airway clearance. Massive or recurrent hemoptysis is treated by bronchial artery embolization or rarely by focal lung resection.
CFTR modulators
CFTR corrector and potentiator medications (especially triple combination therapy) are indicated for approximately 90% of the variants carried by patients with CF. CFTR modulators are not available for patients with class I frameshift and nonsense mutations.
Ivacaftor is a small-molecule oral medication given for maintenance therapy that potentiates the CFTR ion channel in patients with specific CFTR variants. It may be used in patients 1 month of age and older who carry at least 1 copy of a specific variant potentiated by ivacaftor.
Lumacaftor, tezacaftor, and elexacaftor are small-molecule oral medications that partially correct the defective CFTR protein by altering protein misfolding in patients who carry the F508del variant or other specified variants.
The combination of lumacaftor and ivacaftor can be given to patients 1 year of age and older who carry 2 copies of the F508del variant.
The combination of tezacaftor and ivacaftor can be given to patients 6 years of age and older who carry 2 copies of the F508del variant or other specified variants.
The triple combination of elexacaftor, tezacaftor, and ivacaftor can be given to patients 2 years of age and older who carry at least 1 copy of the F508del variant or 1 copy of certain rare variants (4, 5).
Another triple combination of vanzacaftor, tezacaftor, and deuterated ivacaftor is available, which can be given to patients 6 years of age and older who carry at least 1 copy of the F508del variant or 1 copy of certain rare variants (6).
These medications can improve pulmonary function, increase weight, improve exocrine pancreatic function, decrease the frequency of pulmonary exacerbations and hospitalizations, improve quality of life, and reduce and sometimes normalize sweat chloride concentrations (7). The indications for ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor are based on the patient's CFTR variants and age and are subject to change with data available from randomized trials supporting expansion of indications. Although all of these medications can be helpful, ivacaftor alone or in combination with elexacaftor and tezacaftor is considered to be highly effective modulator therapy.
Treatment and prevention of infections
For mild pulmonary exacerbations, a short course of oral antibiotics should be given based on culture and sensitivity testing. The medications of choice for methicillin-sensitive staphylococcus are a penicillinase-resistant penicillin (eg, dicloxacillin), a cephalosporin (eg, cephalexin), or sulfamethoxazole/trimethoprim. Erythromycin, amoxicillin/clavulanic acid, a tetracycline, or linezolid may be used. For patients colonized with methicillin-resistant S. aureus (MRSA), a course of oral sulfamethoxazole/trimethoprim, clindamycin, linezolid, or a tetracycline may be effective. For patients colonized with P. aeruginosa, a short course of inhaled tobramycin or aztreonam (eg, 4 weeks) and/or an oral fluoroquinolone (eg, 2 to 3 weeks) may be effective. Fluoroquinolones may require adjusted dosing but have been used safely in young children (8).
For moderate-to-severe pulmonary exacerbations, especially in patients colonized with P. aeruginosa, IV antibiotic therapy is advised. Patients often require hospital admission, but carefully selected patients can safely receive some of the therapy at home. Combinations of the aminoglycoside tobramycin (or sometimes amikacin) plus a cephalosporin, extended-spectrum penicillin, fluoroquinolone, or monobactam with antipseudomonal activity are given IV, usually for 2 weeks. Higher doses may be required to achieve serum concentrations required for efficacy. Because of enhanced renal clearance in patients with CF, higher doses of some penicillins may be required to achieve adequate serum levels. For patients colonized with MRSA, vancomycin or linezolid can be added to the IV regimen.
Eradication of chronic P. aeruginosa colonization is difficult. It has been shown, however, that early antibiotic treatment around the time the airways are initially infected with P. aeruginosa may be effective in eradicating the organism for some period of time. In patients who are chronically colonized with P. aeruginosa, antibiotics delivered by inhalation (eg, tobramycin) improve clinical parameters and possibly reduce the bacterial burden in the airways (9). The long-term use of alternate-month inhaled tobramycin or aztreonam therapy along with continuous (every month) oral azithromycin given 3 times a week may be effective in improving or stabilizing pulmonary function and decreasing the frequency of pulmonary exacerbations.
Patients who have a clinically significant nontuberculous mycobacterium infection may require long-term therapy with a combination of oral, inhaled, and IV antibiotics.
Treatment of gastrointestinal manifestations
Neonatal intestinal obstruction can sometimes be relieved by enemas containing a hyperosmolar or iso-osmolar radiopaque contrast material; otherwise, surgical enterostomy to flush out the viscid meconium in the intestinal lumen may be necessary. After the neonatal period, episodes of partial intestinal obstruction (distal intestinal obstruction syndrome) can be treated with enemas containing a hyperosmolar or iso-osmolar radiopaque contrast material or acetylcysteine, or by oral administration of a balanced intestinal lavage solution. These enemas generally act by increased intestinal tract volume. If hyperosmolar enemas are used, the high osmolality draws fluid into the intestinal lumen and promotes evacuation. A stool softener such as dioctyl sodium sulfosuccinate (docusate) or lactulose may also help prevent such episodes.
Ursodeoxycholic acid, a hydrophilic bile acid, is often used in patients with liver disease caused by CF, but there is little evidence to support its efficacy in preventing progression from bile stasis to cirrhosis.
Pancreatic enzyme replacement should be given with all meals and snacks to patients with pancreatic insufficiency. The most effective enzyme preparations contain pancrelipase in pH-sensitive, enteric-coated microspheres or microtablets. For infants, the capsules are opened and the contents are mixed with acidic food. After infancy, weight-based dosing is used. High doses should be avoided because they have been associated with fibrosing colonopathy. In patients with high enzyme requirements, acid suppression with an H2 blocker or proton pump inhibitor may improve enzyme effectiveness.
Diet therapy includes sufficient calories and protein to promote normal growth—30 to 50% higher than the usual recommended dietary allowances may be required to offset the increased energy expenditures associated with tachypnea, malabsorption, and a chronic inflammatory state associated with heightened immune responsiveness (see table ). Diet therapy also includes a normal-to-high total fat intake to increase the caloric density of the diet, a water-miscible multivitamin supplement in double the recommended daily allowance, supplementation with vitamin D3 (cholecalciferol) in patients with vitamin D deficiency or insufficiency, and salt supplementation during infancy and periods of thermal stress and increased sweating. Infants receiving broad-spectrum antibiotics and patients with liver disease and hemoptysis should be given additional supplemental vitamin K. Formulas containing protein hydrolysates and medium-chain triglycerides may be used instead of modified whole-milk formulas for infants with severe malabsorption. Glucose polymers and medium-chain triglyceride supplements can also be used to increase caloric intake.
In patients who fail to maintain adequate nutritional status, enteral supplementation via gastrostomy or jejunostomy may improve growth and stabilize pulmonary function (see Overview of Nutritional Support). The use of appetite stimulants to enhance growth may be helpful in some patients.
Treatment of other manifestations
Cystic fibrosis–related diabetes (CFRD) is caused by insulin insufficiency and shares features of both type 1 and type 2 diabetes. Insulin is the only recommended treatment. Management includes an insulin regimen, nutrition counseling, a diabetes self-management education program, and monitoring for microvascular complications. The management plan should be carried out in conjunction with an endocrinologist and a dietitian with experience in treating both CF and diabetes.
Patients with symptomatic right heart failure should be treated with diuretics, salt restriction, and oxygen.
Recombinant human growth hormone (rhGH) is rarely used but may improve pulmonary function, increase height and weight and bone mineral content, and reduce the rate of hospitalization.
Surgery may be indicated for localized bronchiectasis or atelectasis that cannot be treated effectively with medications, nasal polyps, chronic rhinosinusitis, bleeding from esophageal varices secondary to portal hypertension, gallbladder disease, and intestinal obstruction due to a volvulus or an intussusception that cannot be medically reduced.
Liver transplantation has been done successfully in patients with end-stage liver disease.
Often, discussion of lung transplantation is needed. In considering transplantation, patients need to weigh the merits of longer survival with a transplant against the uncertainty of getting a transplant and the ongoing (but different) burden of living with an organ transplant. Bilateral cadaveric lung transplantation and live donor lobar transplantation have been done successfully in patients with advanced pulmonary disease. Combined liver-lung transplantation has been done for patients with end-stage liver and lung disease.
Bilateral lung transplantation should be a consideration for patients with severe lung disease. Among adults with CF, median survival posttransplant is 9.5 years (10).
Treatment references
1. Flume PA, O'Sullivan BP, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007;176(10):957-969. doi:10.1164/rccm.200705-664OC
2. Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2013;187(7):680-689. doi:10.1164/rccm.201207-1160oe
3. Stahl M, Wielpütz MO, Ricklefs I, et al. Preventive Inhalation of Hypertonic Saline in Infants with Cystic Fibrosis (PRESIS). A Randomized, Double-Blind, Controlled Study. Am J Respir Crit Care Med 2019;199(10):1238-1248. doi:10.1164/rccm.201807-1203OC
4. Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial [published correction appears in Lancet 2020 May 30;395(10238):1694]. Lancet 2019;394(10212):1940-1948. doi:10.1016/S0140-6736(19)32597-8
5. Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 2019;381(19):1809-1819. doi:10.1056/NEJMoa1908639
6. Keating C, Yonker LM, Vermeulen F, et al. Vanzacaftor-tezacaftor-deutivacaftor versus elexacaftor-tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years and older (SKYLINE Trials VX20-121-102 and VX20-121-103): results from two randomised, active-controlled, phase 3 trials. Lancet Respir Med 2025;13(3):256-271. doi:10.1016/S2213-2600(24)00411-9
7. Taylor-Cousar JL, Robinson PD, Shteinberg M, Downey DG. CFTR modulator therapy: transforming the landscape of clinical care in cystic fibrosis. Lancet 2023;402(10408):1171-1184. doi:10.1016/S0140-6736(23)01609-4
8. Patel K, Goldman JL. Safety Concerns Surrounding Quinolone Use in Children. J Clin Pharmacol 2016;56(9):1060-1075. doi:10.1002/jcph.715
9. Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic Fibrosis Foundation pulmonary guideline. pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann Am Thorac Soc 2014;11(10):1640-1650. doi:10.1513/AnnalsATS.201404-166OC
10. Chambers DC, Cherikh WS, Goldfarb SB, et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-fifth adult lung and heart-lung transplant report-2018; Focus theme: Multiorgan Transplantation. J Heart Lung Transplant 2018;37(10):1169-1183. doi:10.1016/j.healun.2018.07.020
Prognosis for Cystic Fibrosis
The course is largely determined by the degree of pulmonary involvement. Deterioration of pulmonary function over time, generally characterized by progressive bronchiectasis, leads to debilitation and eventually increases the risk for death, usually due to a combination of respiratory failure and cor pulmonale.
Prognosis has improved steadily over the past 5 decades, mainly because of early diagnosis and aggressive treatment before the onset of irreversible pulmonary changes. Median age at death in 2023 was 36.9 years. However, median predicted survival in the United States for children born in 2023 is age 68 years. Long-term survival is significantly better in patients without pancreatic insufficiency (1). Outcomes are also affected by CFTR variant profile, modifier genes, airway microbiology, sex, ambient temperature, exposure to air pollutants (including tobacco smoke), adherence to prescribed therapies, and socioeconomic status. The FEV1, adjusted for age and sex, is the best predictor of survival. If health outcomes with CFTR modulator therapy are sustained, life expectancy can potentially increase even further.
End-of-life care
Patients and their families should have empathetic discussions of prognosis and preferences for care throughout the course of illness, especially if pulmonary function progressively declines (2).
Patients with CF should have adequate access to information and opportunities to make important life choices, including active participating in end-of-life decision making.
When appropriate, palliative care, including sufficient symptom management, should be offered to ensure peaceful end-of-life care. A useful strategy for patients to consider is to accept a time-limited trial of fully aggressive treatment when needed, but to agree in advance to parameters that indicate when to stop aggressive measures (see Do-Not-Resuscitate (DNR) Orders and Portable Medical Orders).
Prognosis references
1. Cystic Fibrosis Foundation. Understanding Changes in Life Expectancy. Accessed July 31, 2025.
2. Kavalieratos D, Georgiopoulos AM, Dhingra L, et al. Models of Palliative Care Delivery for Individuals with Cystic Fibrosis: Cystic Fibrosis Foundation Evidence-Informed Consensus Guidelines. J Palliat Med 2021;24(1):18-30. doi:10.1089/jpm.2020.0311
Key Points
Cystic fibrosis is caused by carrying 2 variants of the gene for a protein called the cystic fibrosis transmembrane conductance regulator (CFTR), which regulates chloride, sodium, and bicarbonate transport across epithelial membranes.
The main complications involve the lungs, with damage to the small and large airways, chronic inflammation, and chronic and recurrent bacterial infections, particularly by Pseudomonas aeruginosa.
Other major consequences include pancreatic insufficiency, leading to malabsorption of nutrients and vitamins with consequent impaired growth and development, and, in older patients, a risk for developing diabetes.
Airway clearance measures (eg, postural drainage, percussion, vibration, assisted coughing), mucolytics, and airway hydrators are often started in early childhood; regular aerobic exercise is recommended.
Medications that correct or potentiate CFTR (CFTR modulators) can improve health outcomes for patients who have certain CFTR variants.
Antibiotics are given early in any pulmonary exacerbation; medication selection may be based on culture and sensitivity testing.
Diet should be supplemented with pancreatic enzymes, high-dose vitamins, and 30 to 50% more calories derived primarily from fat.
More Information
The following English-language resources may be useful. Please note that The Manual is not responsible for the content of these resources.
Cystic Fibrosis Foundation: Age-specific care guidelines for cystic fibrosis
Cystic Fibrosis Foundation: Clinical Care Guidelines
Green DM, Lahiri T, Raraigh KS, et al. Cystic Fibrosis Foundation Evidence-Based Guideline for the Management of CRMS/CFSPID. Pediatrics 2024;153(5):e2023064657. doi:10.1542/peds.2023-064657
Drug Information for the Topic


