


Primary malignant bone tumors are much less common than metastatic bone tumors, particularly in adults. Primary malignant bone tumors include multiple myeloma, osteosarcoma, adamantinoma, chondrosarcoma, chordoma, Ewing sarcoma of bone, fibrosarcoma and undifferentiated pleomorphic sarcoma, lymphoma of bone, and malignant giant cell tumor. (See also Overview of Bone and Joint Tumors and Overview of Leukemia.)
The two most widely used systems for staging these tumors are
The American Joint Committee on Cancer (AJCC) Cancer Staging Manual, 8th edition: For osteosarcoma, chondrosarcoma, and Ewing sarcoma, staging is based on distinct tumor category, histologic grade, size, nodal involvement, and metastases (TNM classification). The manual classifies tumors into 4 stages and is used for reporting cancer data.
The Musculoskeletal Tumor Society (MSTS) staging system: Used by orthopedic oncology surgeons based on histologic grade (eg, Stage I-low-grade histology and Stage II-high-grade histology, whether the tumor is contained entirely within the bone (A) or has broken outside of the cortex into surrounding soft tissue (B), and metastases Stage III). The typical (conventional) osteosarcoma with an associated soft tissue mass without metastases is Stage IIB in the MSTS system.
Multiple myeloma
Multiple myeloma is the most common primary malignant bone tumor but is often considered a marrow cell tumor within the bone rather than a primary bone tumor because it is of hematopoietic derivation (see also Multiple Myeloma). Even if multiple myeloma is considered a hematologic tumor, the identified skeletal abnormality must be differentiated from other bone tumors.
Multiple myeloma occurs mostly in older adults.
Tumor development and progression is usually multicentric and often involves the bone marrow so diffusely that bone marrow aspiration is diagnostic. Unlike in metastatic disease, a radionuclide bone scan may not reliably show lesions and skeletal surveys should be done. Skeletal surveys typically show sharply circumscribed lytic lesions (punched-out lesions) or diffuse demineralization. Rarely, the lesion can appear as sclerotic or as diffuse osteopenia, especially in a vertebral body. An isolated single myeloma lesion without systemic marrow involvement is called a plasmacytoma.

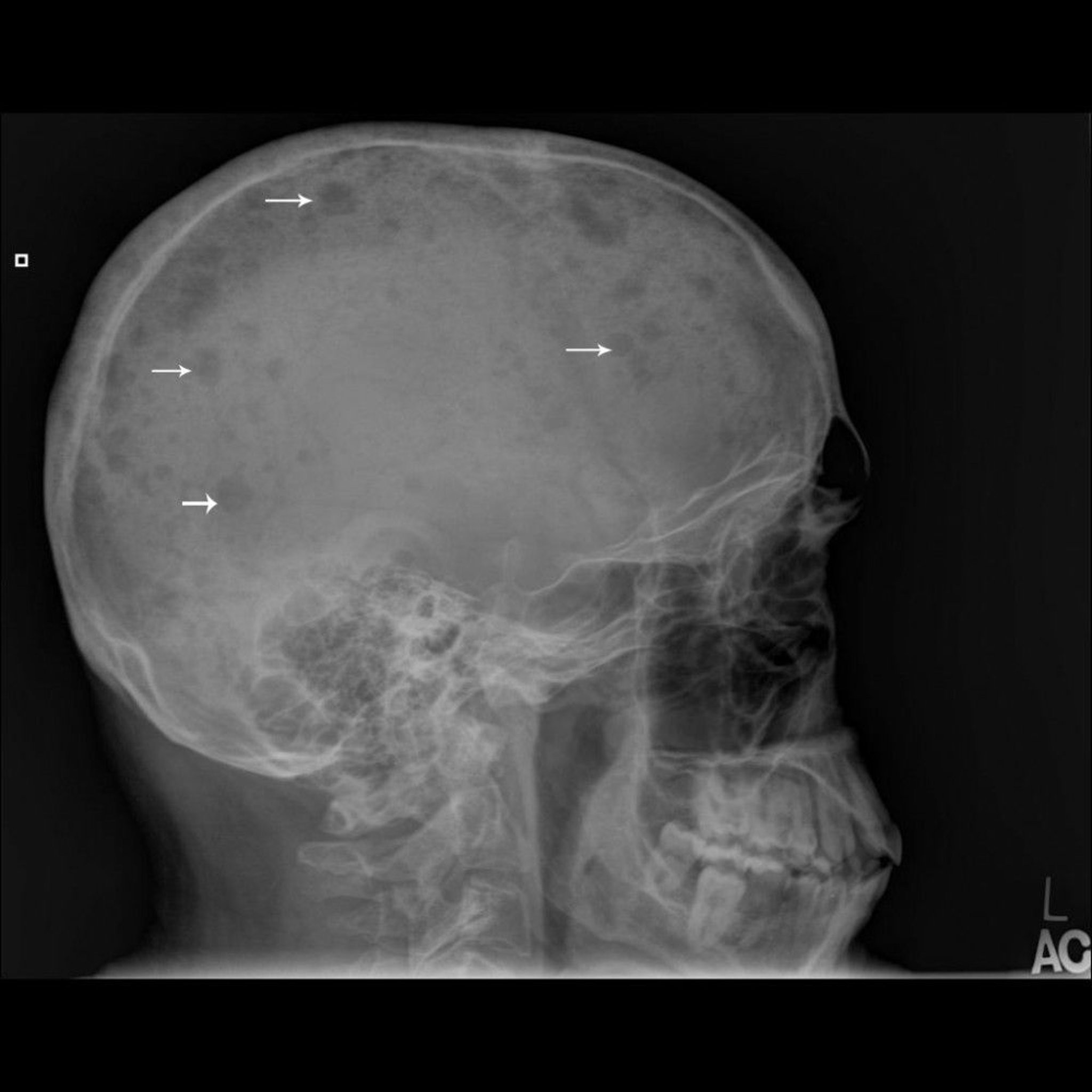
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Certain bony lesions respond quite well to radiation therapy.
Osteosarcoma (osteogenic sarcoma)
Osteosarcoma is the most common malignant primary bone tumor (if one considers myeloma a marrow cell tumor and not a primary bone tumor) and is highly malignant. It is most common among people aged 10 to 25, although it can occur at any age. There are two peaks of incidence; incidence is highest in adolescents and very young adults (coinciding with adolescent growth spurt) and secondary peak occurs in older adults (≥ age 60), especially in those with risk factors such as Paget disease, bone infarcts, and areas of bone previously exposed to high-dose radiation therapy for another cancer many years earlier. There is a genetic predisposition, especially in children who carry the gene for hereditary retinoblastoma (variants of RB1 gene) and Li-Fraumeni syndrome (TP53 gene).
Osteosarcoma produces malignant osteoid (immature bone) from tumor bone cells. Osteosarcoma usually develops around the knee (distal femur more often than proximal tibia) or in other long bones, particularly the metaphyseal-diaphyseal area, and may metastasize, usually to lung or other bone. Pain and swelling are the usual symptoms.

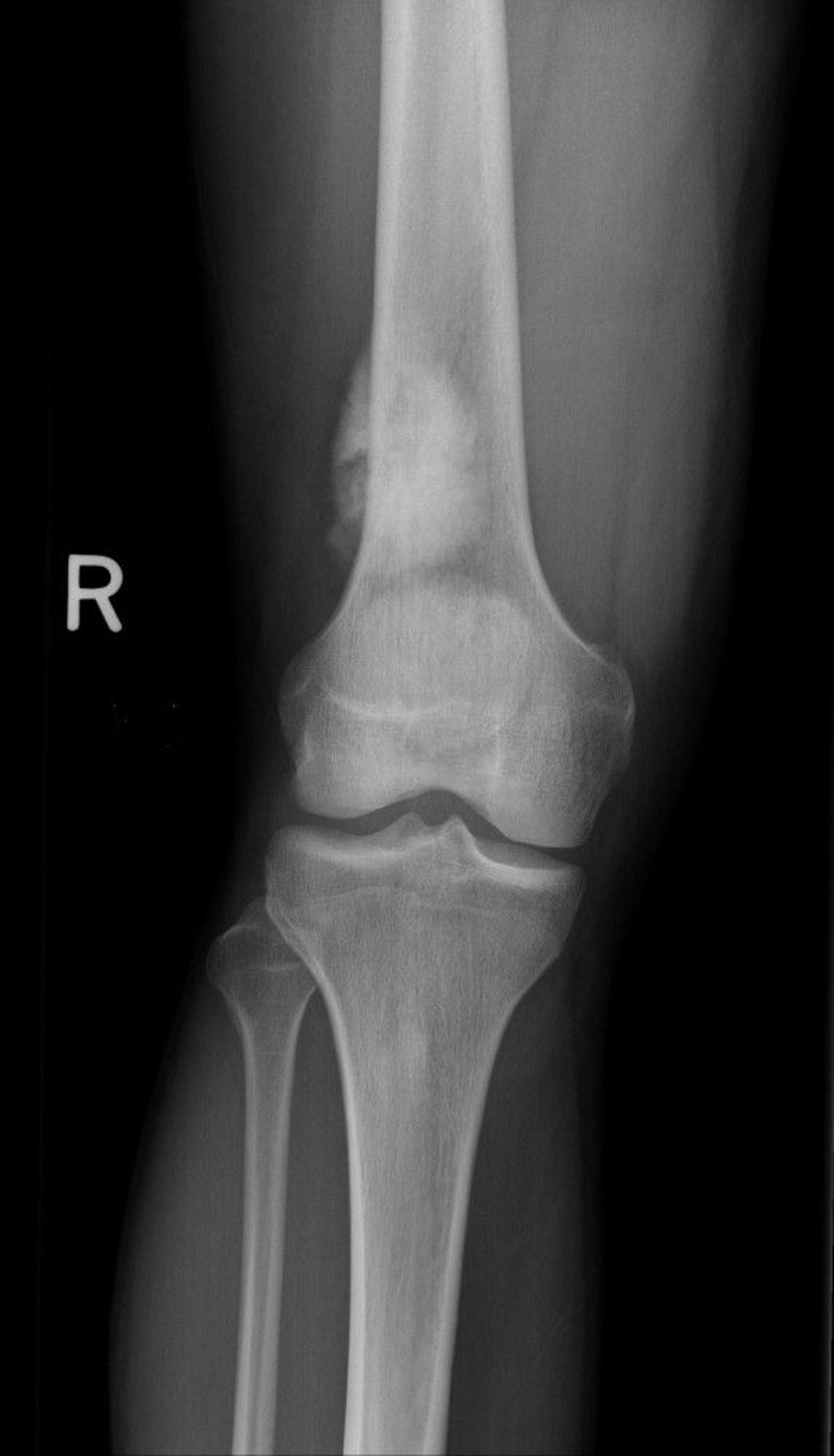
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Findings on imaging studies vary and may include sclerotic or lytic features. Diagnosis of osteosarcoma requires biopsy. Patients need a chest x-ray and CT to detect lung metastases and a bone scan to detect bone metastases. MRI is done of the entire involved extremity to detect metachronous lesions if present. PET-CT may show distant metastases or metachronous lesions.
Treatment of osteosarcoma is a combination of chemotherapy and surgery. Use of adjuvant chemotherapy increases survival from < 20% to > 65% at 5 years. Neoadjuvant chemotherapy begins before surgical resection. Decreased peripheral soft tissue tumor mass or increased mineralization on radiographs decreased pain level, and decreased serum alkaline phosphatase indicate some response, but the desired response is for > 95% tumor necrosis on histologic mapping of the resected specimen by the pathologist. After several courses of chemotherapy (over several months), limb-sparing surgery and limb reconstruction can proceed. On occasion, a surgical amputation is done before the start of chemotherapy for a fungating tumor. The goal is to treat the early micrometastatic disease assumed to be present even if not seen on staging imaging studies.
In limb-sparing surgery, the tumor is resected en bloc, including all surrounding reactive tissue and a rim of surrounding normal tissue; to avoid microscopic spillage of tumor cells, the tumor is not violated. More than 85% of patients can be treated with limb-sparing surgery without decreasing the long-term survival rate.
Continuation of chemotherapy after surgery is necessary. If there is nearly complete tumor necrosis (about 95%) from preoperative chemotherapy, 5-year survival rate is > 90%. Limited metastatic disease to the lungs sometimes may be treated with thoracotomy and wedge resection of the lung lesion(s).
Variants of osteosarcoma that are different from conventional osteosarcoma and occur much less frequently include surface cortical lesions, such as parosteal osteosarcoma and periosteal osteosarcoma. Parosteal osteosarcomas most often involve the posterior cortex of the distal femur and usually are fairly well-differentiated. Chemotherapy is not required before surgical resection for the treatment of low-grade parosteal osteosarcoma. Parosteal osteosarcomas require surgical en bloc resection but no chemotherapy if histology of the resected specimen confirms the tumor to be well-differentiated.
Periosteal osteosarcoma is more of a cartilage matrix surface tumor that also contains bone matrix and is malignant. It is often located on the mid-shaft femur and appears as a sunburst on x-ray. Likelihood of metastases for periosteal osteosarcomas is much greater than for well-differentiated parosteal osteosarcomas, but somewhat less than for typical osteosarcomas. Most of the time, periosteal osteosarcomas are treated similarly to conventional osteosarcomas with chemotherapy and surgical en bloc resection.
Adamantinoma
Adamantinoma is rare (< 1% of malignant bone tumors) and most often develops in the tibia. It usually occurs in adolescents and people who are in their 20s but can occur at any age. Adamantinoma is slow-growing and often manifests with pain and palpable fullness.

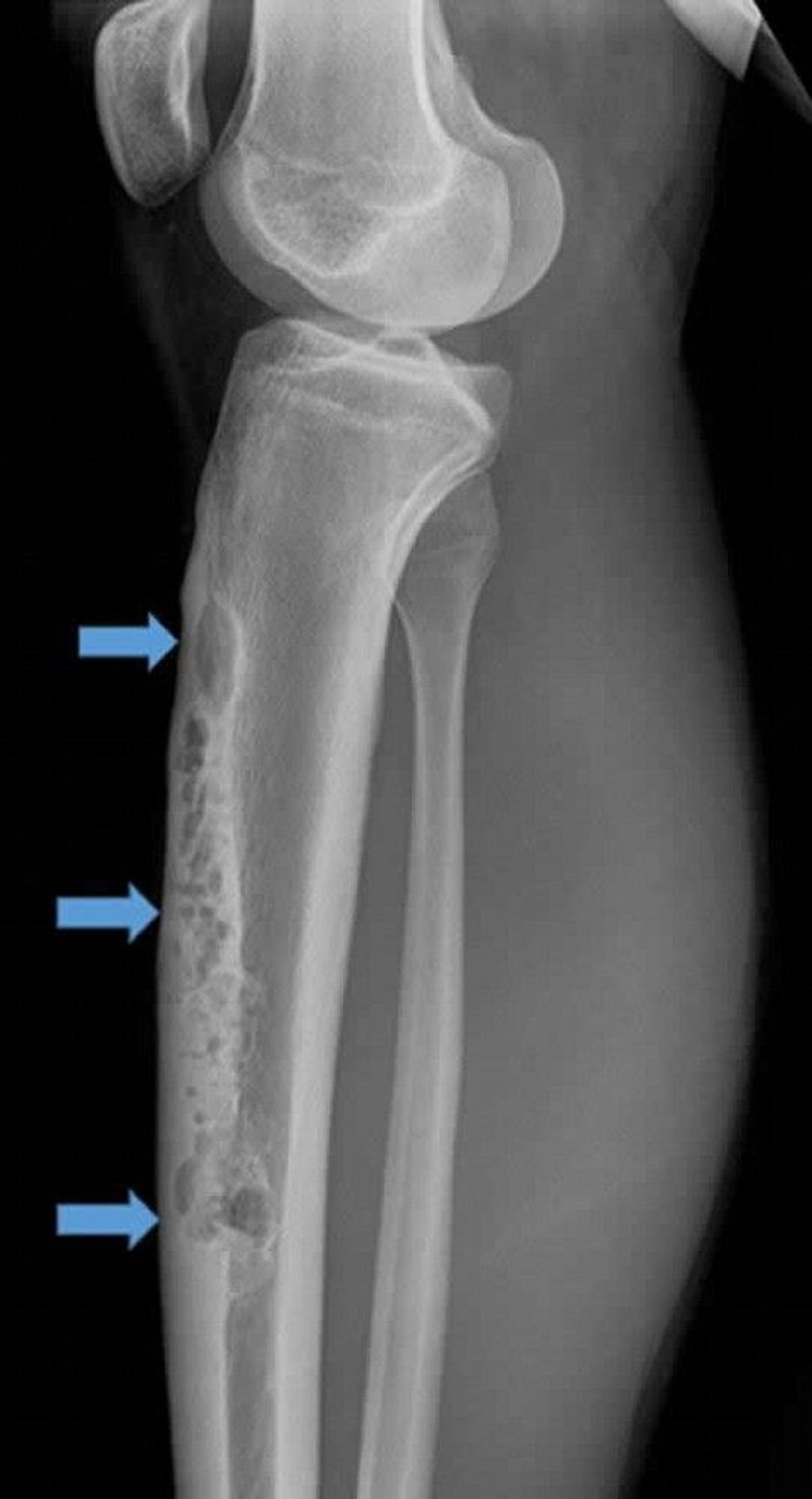
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
The lesion typically manifests in the anterior crest of the tibia, and x-rays show a "soap bubble" osteolytic appearance. The histologic appearance is a biphasic pattern of epithelial and osteofibrous tissue. The lesion can be confused with osteofibrous dysplasia of the anterior tibial cortex, which is benign. Some clinicians think osteofibrous dysplasia of the anterior tibial cortex may be a precursor to adamantinoma but without the epithelial component that would then make it a cancer.
Metastases do occur, primarily to the lungs, but are rare.
Treatment of adamantinoma consists of wide resection and reconstruction of the defect. Occasionally, amputation is necessary.
Chondrosarcoma
Chondrosarcomas are malignant tumors of cartilage. They differ from osteosarcomas clinically, therapeutically, and prognostically. Of chondrosarcomas, 90% are primary tumors. Chondrosarcomas can also arise in other preexisting conditions, particularly multiple osteochondromas and multiple enchondromatosis (eg, in Ollier disease and Maffucci syndrome). Chondrosarcomas tend to occur in older adults. They often develop in flat bones (eg, pelvis, scapula) but can develop in any portion of any bone (most often femur and humerus among the long bones) and can have a soft tissue tumor component involving surrounding soft tissues.

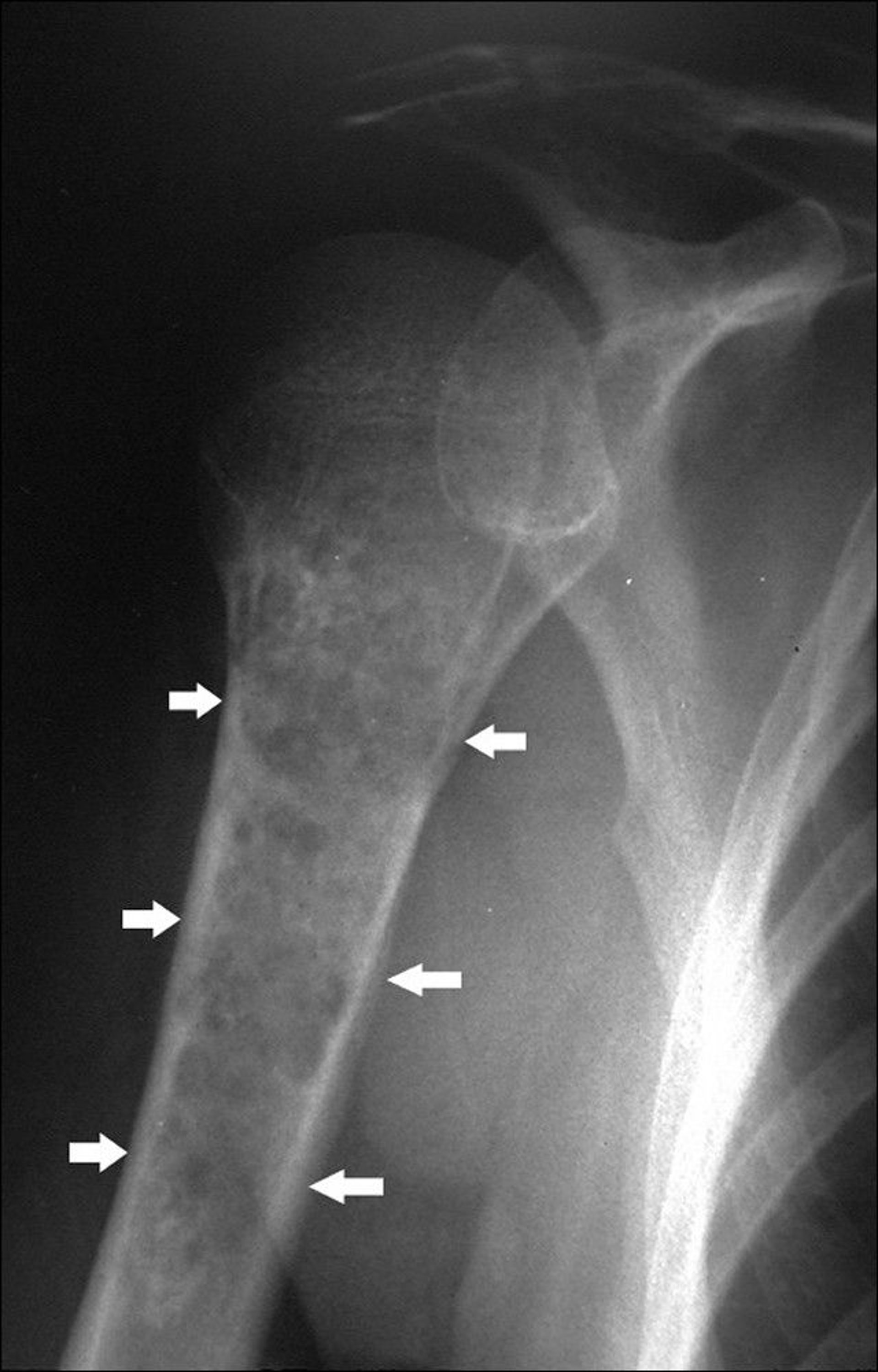
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
X-rays often reveal punctate calcifications. Chondrosarcomas often also exhibit cortical bone destruction and loss of normal bone trabeculae. MRI may show a soft tissue mass. Bone scan may also be done. Tissue diagnosis is required for chondrosarcoma and can also determine the tumor’s grade (probability of metastasizing). Needle biopsy may provide an inadequate tissue sample.
It is often difficult to differentiate low-grade chondrosarcomas from enchondromas by imaging and sometimes even histology.
Chordoma
Chordoma, which is rare, develops from the remnants of the primitive notochord. It tends to occur at the ends of the spinal column, usually in the middle of the sacrum or near the base of the skull. A chordoma in the sacrococcygeal region causes nearly constant pain. A chordoma in the base of the skull can cause deficits in a cranial nerve, most commonly in nerves to the eye.
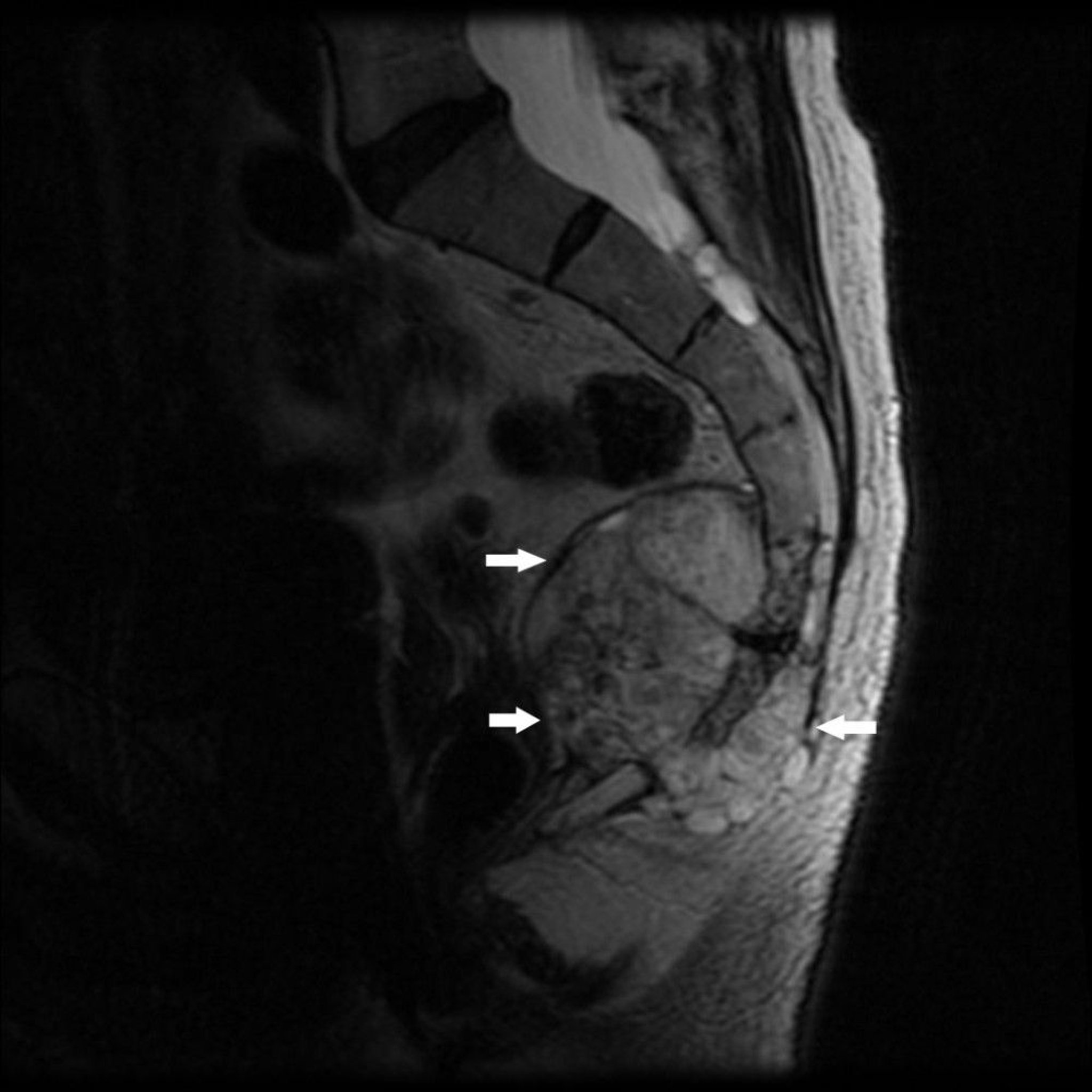
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Symptoms of chordoma may exist for months to several years before diagnosis.
A chordoma appears on imaging studies as a destructive bone lesion that may be associated with a soft tissue mass. Biopsy is done.
Chordomas in the sacrococcygeal region may be cured by radical en bloc excision. Chordomas in the base of the skull are usually inaccessible to surgery but may respond to radiation therapy. The rate of local recurrence is high after tumor resection. Metastasis, although less common, may occur.
Ewing sarcoma of bone
Ewing sarcoma of bone is a small blue round-cell bone tumor with a peak incidence between 10 and 20 years. Ewing sarcoma is related to peripheral primitive neuroectodermal tumors (PNET) and Askin malignant small cell tumor of the chest wall, which are now considered part of the Ewing sarcoma family. Most tumors develop in the extremities, but any bone may be involved. Ewing sarcoma tends to be extensive, sometimes involving the entire bone shaft, most often the diaphyseal region. About 15 to 20% occur around the metaphyseal region. Pain and swelling are the most common symptoms.

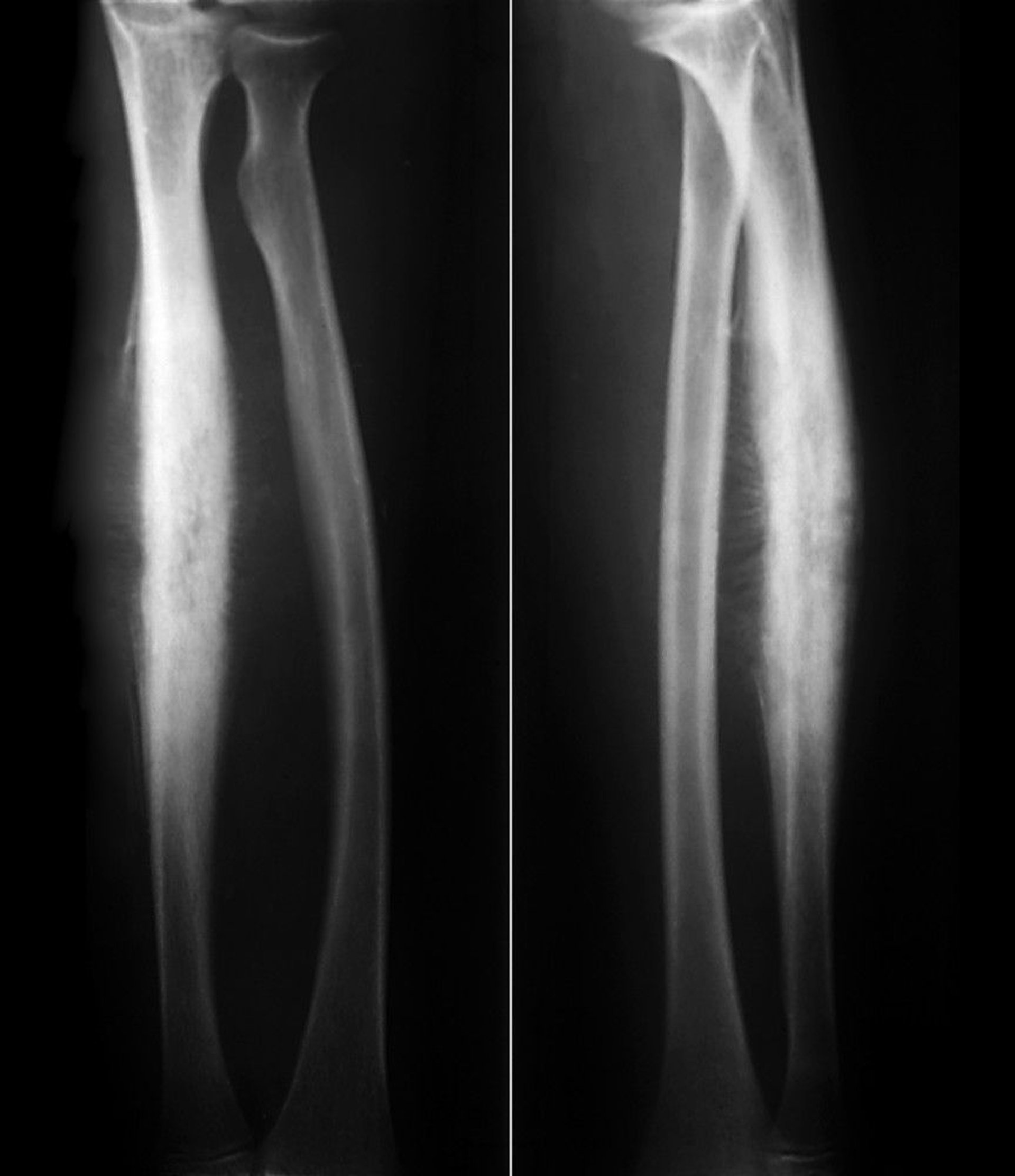
ZEPHYR/SCIENCE PHOTO LIBRARY
Lytic destruction, particularly a permeative infiltrating pattern without clear borders, is the most common finding on imaging, but multiple layers of subperiosteal reactive new bone formation may give an onion-skin appearance. X-rays do not usually reveal the full extent of bone involvement, and a large soft tissue mass usually surrounds the affected bone. MRI better defines disease extent, which can help guide treatment.
Many other benign and malignant tumors can appear very similarly, so diagnosis of Ewing sarcoma is made by biopsy. At times this type of tumor may be confused with an infection. Accurate histologic diagnosis can be accomplished with cytogenetic analysis and identification of molecular markers, including evaluation for a typical clonal chromosomal abnormality. Molecular findings in the Ewing family of tumors are distinct nonrandom chromosomal translocations involving the Ewing sarcoma gene (EWS) on chromosome 22. Eighteen different structural translocations in different fusion gene patterns have been identified.

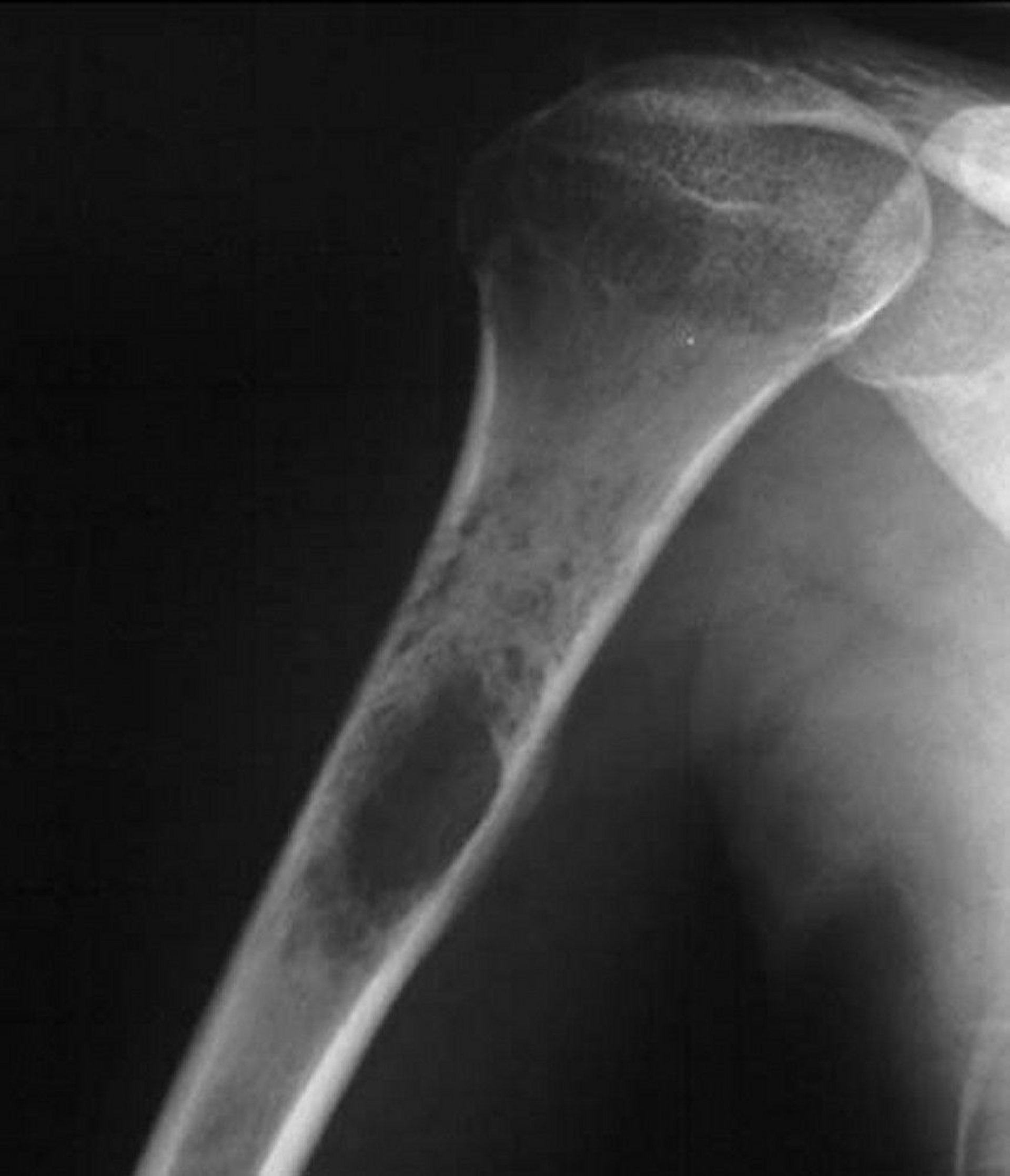
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Treatment of Ewing sarcoma includes various combinations of surgery, chemotherapy, and radiation therapy. Currently, > 60% of patients with primary localized Ewing sarcoma may be cured by this multimodal approach. Cure is sometimes possible even with metastatic disease. Chemotherapy in conjunction with surgical en bloc resection, if applicable, often yields better long-term results than chemotherapy in conjunction with radiation therapy.
Fibrosarcoma and undifferentiated pleomorphic sarcoma (formerly malignant fibrous histiocytoma of bone)
Fibrosarcomas and undifferentiated pleomorphic sarcoma have similar characteristics to osteosarcomas but produce fibrous tumor cells (rather than bone tumor cells), affect the same age group, and pose similar problems.
Treatment and outcome for high-grade lesions are similar to osteosarcoma.
Lymphoma of bone
Lymphoma of bone (previously known as reticulum cell sarcoma) affects adults, usually in their 40s and 50s. This usually is a diffuse large B-cell lymphoma. It may arise in any bone. The tumor consists of small round cells, often with a mixture of reticulum cells, lymphoblasts, and lymphocytes. It can develop as an isolated primary bone tumor, in association with similar tumors in other tissues, or as a metastasis from known soft tissue lymphomatous disease. Pain and swelling are the usual symptoms of lymphoma of bone. Pathologic fracture is common.

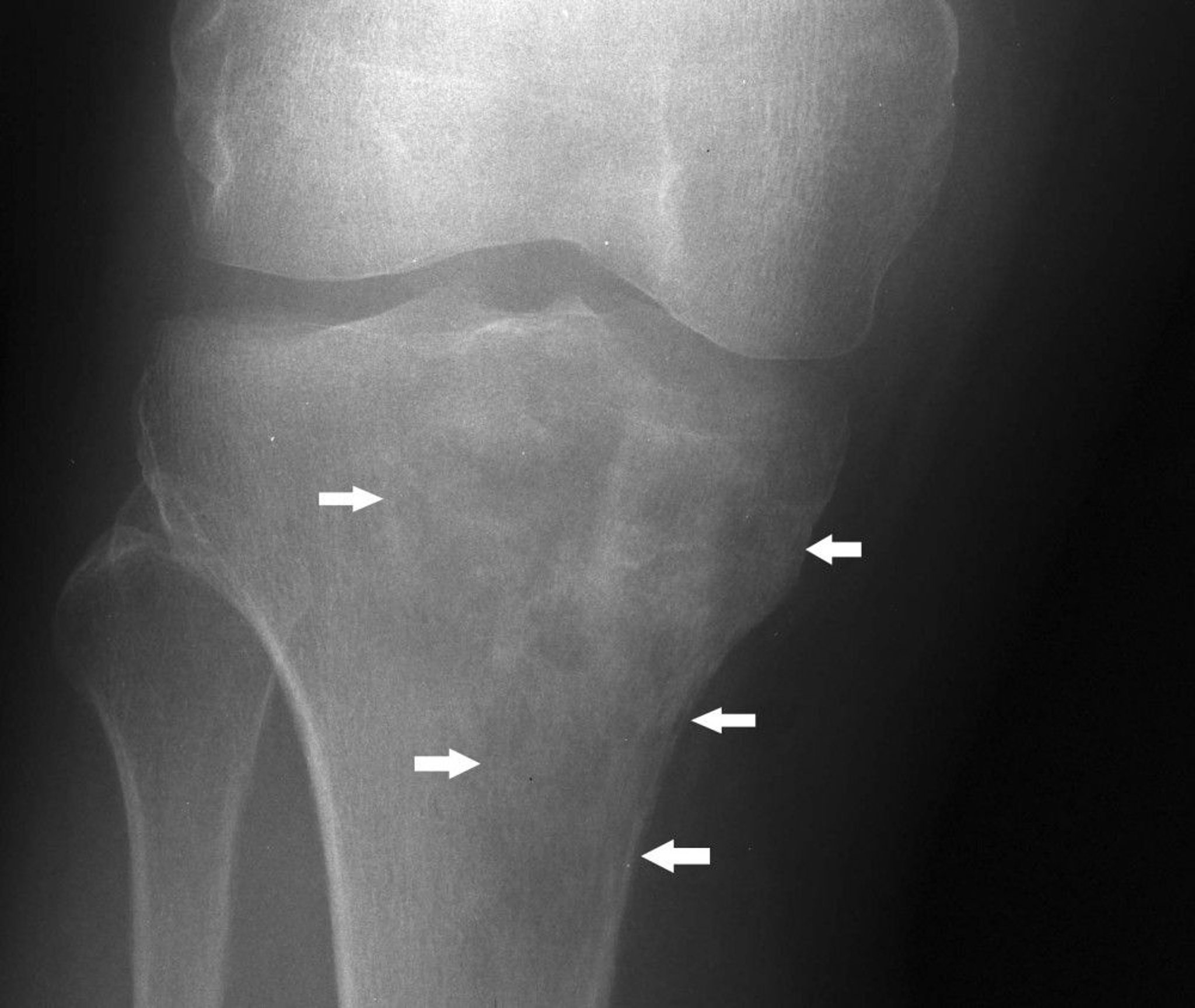
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Imaging studies reveal bone destruction, which may be in a mottled or patchy or even infiltrating, permeative pattern, often with a clinical and radiographic large soft tissue mass. In advanced disease, the entire outline of the affected bone may be lost. Biopsy is also done.
In isolated primary bone lymphoma, the 5-year survival rate is ≥ 50%.
Bone lymphomas are typically treated with systemic chemotherapy. Radiation therapy can be used as an adjuvant in some cases. Stabilization of long bones is often necessary to prevent pathologic fracture. Amputation is indicated only rarely, when function is lost because of pathologic fracture or extensive soft tissue involvement that cannot be managed otherwise.
Malignant giant cell tumor
Malignant giant cell tumor, which is rare, is usually located at the extreme end of a long bone.
X-ray reveals classic features of malignant destruction (predominantly lytic destruction, cortical destruction, soft tissue extension, and pathologic fracture). A malignant giant cell tumor that develops in a previously benign giant cell tumor is characteristically radioresistant. MRI and biopsy are done.
Treatment of malignant giant cell tumor is similar to that of osteosarcoma, but the cure rate is low.






