

Cystic fibrosis is a hereditary disease that causes certain glands to produce abnormally thick secretions, resulting in tissue and organ damage, especially in the lungs and the digestive tract.
Cystic fibrosis is caused by inherited abnormal genes changing the function of the cystic fibrosis protein leading to thick, sticky secretions to clog the lungs and other organs.
Typical symptoms include coughing, wheezing, and frequent respiratory tract infections throughout life as well as abdominal bloating, loose stools, and poor weight gain.
The diagnosis is based on sweat test results and/or genetic testing.
More than half of the people with this disease in the United States are adults.
Treatments include antibiotics, bronchodilators, medications to thin lung secretions, airway clearance treatments for respiratory problems, supplements of pancreatic enzymes and vitamins for digestive problems, and medications to improve the function of the cystic fibrosis protein in people with certain abnormal genes.
Some people may require liver and lung transplantation.
Cystic fibrosis is an inherited disease that leads to a shortened life span. In the United States, it occurs in about 1 of 3,300 White infants and in 1 of 15,300 Black infants. It is rare in people of Asian descent. There are approximately 40,000 people with cystic fibrosis living in the United States, and approximately 105,000 diagnosed with cystic fibrosis worldwide. Because improvements in treatment have extended life expectancy for people with cystic fibrosis, almost 60 percent of the people in the United States with this disease are adults. Cystic fibrosis is equally common among boys and girls.


Section A of the illustration shows the organs that may be affected by cystic fibrosis (sinuses, lungs, skin, liver, pancreas, intestines, reproductive organs).
Section B shows a normal airway with a thin layer of mucus lining the wall.
Section C shows an airway with cystic fibrosis. The widened airway is blocked by thick, sticky mucus that contains blood and bacteria.
Source: National Heart, Lung, and Blood Institute; National Institutes of Health; U.S. Department of Health and Human Services.
Causes of Cystic Fibrosis
Abnormal genes
Cystic fibrosis results when a person inherits 2 defective copies (variants) of a particular gene, one from each parent. This gene is called the cystic fibrosis transmembrane conductance regulator (CFTR). There are a number of variants of the CFTR gene. For example, the most common one is called the F508del variant. The CFTR gene controls the production of a protein that regulates the movement of chloride, bicarbonate, and sodium (salt) across cell membranes. Variants of the CFTR gene cause the protein to become dysfunctional. If the protein does not work correctly, the movement of chloride, bicarbonate, and sodium is disrupted, leading to thickening and increased stickiness of secretions throughout the body.
Worldwide, about 4% of White people carry one defective copy of the CFTR gene. People with one defective copy are carriers, but they themselves do not get sick. When people inherit 2 defective copies of the gene, they develop cystic fibrosis.
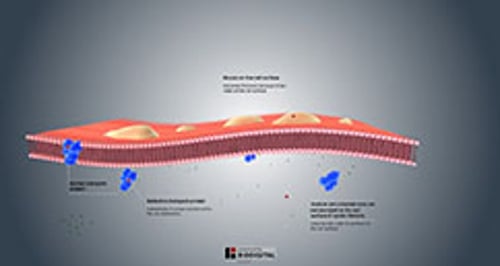
Abnormal secretions
Cystic fibrosis affects many organs throughout the body and nearly all the glands that secrete fluids into a duct (exocrine glands).
The organs most commonly affected are the:
Lungs
Pancreas
Intestines
Liver and gallbladder
Reproductive organs
The lungs are normal at birth, but problems can develop at any time afterward as thick secretions begin to block the small airways (mucus plugging). The plugging leads to chronic bacterial infections and inflammation that cause permanent damage to the airways (bronchiectasis). These problems make breathing increasingly difficult and reduce the lungs’ ability to transfer oxygen to the blood. People also may have frequent bacterial respiratory infections that affect the sinuses.
In the pancreas, blockage of ducts prevents digestive enzymes from reaching the small intestine. A lack of these enzymes leads to poor absorption of fats, proteins, and vitamins (malabsorption). This poor absorption, in turn, can lead to nutritional deficiencies and poor growth. Eventually, the pancreas can become scarred and no longer produce enough insulin, so some people develop diabetes. However, a small percentage of people who have cystic fibrosis and carry certain variants do not develop pancreatic digestive problems.
The intestines can become blocked by thick secretions. This blockage is common immediately after birth because the contents of the fetus's digestive tract (called meconium) are abnormally thick. Such blockage in the small intestine is called meconium ileus and in the large intestine is called meconium plug syndrome. Older children and adults may also have problems with constipation and blockage of the intestines (distal intestinal obstruction syndrome).
The liver and gallbladder can be blocked by thick secretions, which eventually can cause liver scarring (fibrosis). Gallstones may develop.
The reproductive organs can be blocked by thick secretions, which can cause infertility. Almost all men are infertile, but infertility is much less common in women.
The sweat glands secrete fluid containing more salt than normal, increasing the risk of dehydration.
Symptoms of Cystic Fibrosis
The symptoms of cystic fibrosis can vary depending on a person's age.
Newborns and young children
About 20% of newborns who have cystic fibrosis have meconium ileus, which causes vomiting, bloating (distention) of the abdomen, and absence of bowel movements. Meconium ileus is sometimes complicated by perforation of the intestine, a dangerous condition causing infection and peritonitis (inflammation of the tissue lining the abdominal cavity and abdominal organs) and, if untreated, shock and death. Some newborns have a twisting of the intestine on itself (volvulus) or incomplete development of the intestine. Newborns who have meconium ileus almost always develop other symptoms of cystic fibrosis.
The first symptom of cystic fibrosis in an infant who does not have meconium ileus is often a delay in regaining birth weight or poor weight gain at 4 to 6 weeks of age. This poor weight gain is due to poor absorption of nutrients related to inadequate amounts of pancreatic enzymes. The infant has frequent, bulky, foul-smelling, oily stools (called steatorrhea) and may have a bloated (distended) abdomen. Without treatment, weight gain is slow despite a normal or large appetite.
Older children and adults
Unless a diagnosis is made through newborn screening, about half the children with cystic fibrosis are first taken to the doctor because of frequent coughing, wheezing, and respiratory tract infections. Coughing, the most noticeable symptom, is often accompanied by gagging, vomiting, and disturbed sleep. Children may have difficulty breathing, wheezing, or both. As the disease progresses, children develop a declining tolerance for exercise, lung infections tend to occur more frequently, the chest becomes barrel-shaped, and insufficient oxygen may make the fingers clubbed (see Clubbing) and the nail beds bluish. Polyps may form in the nose. The sinuses may fill with thick secretions and become infected, leading to chronic or recurring sinusitis.
Older children and adults may have episodes of constipation or develop recurring and sometimes chronic blockage of the intestines. Symptoms include a change in stooling pattern, crampy abdominal pain, a decreased appetite, and sometimes vomiting. Gastroesophageal reflux is relatively common among children and adults.
When a child or adult with cystic fibrosis sweats excessively in hot weather or because of a fever, dehydration may result because of the increased loss of salt and water. Parents may notice the formation of salt crystals or even a salty taste on their child’s skin.
Adolescents often have slowed growth and delayed puberty.
As the disease progresses, recurrent lung infection become a major concern and cause lasting damage to the lungs.
Complications of Cystic Fibrosis
There are many complications of cystic fibrosis.
Inadequate absorption of the fat-soluble vitamins A, D, E, and K may lead to night blindness, osteopenia (decrease in bone density), osteoporosis, anemia, and bleeding disorders. In untreated infants and toddlers, the lining of the rectum may protrude through the anus, a condition called rectal prolapse. Infrequently, infants with cystic fibrosis who have been fed soy formula or hypoallergenic formula may develop anemia and swelling of the extremities, because they are not absorbing enough protein.
Complications of advanced cystic fibrosis in adolescents and adults include rupture of the small air sacs of the lung (alveoli) into the pleural space (the space between the lung and chest wall). This rupture can allow air to enter into the pleural space (pneumothorax), which collapses the lung. Other complications include heart failure and massive or recurring bleeding in the airways.
About 20% of adolescents and up to 50% of adults with cystic fibrosis develop diabetes for which they must take insulin because the scarred pancreas can no longer produce enough insulin.
The blockage of bile ducts by thick secretions can lead to inflammation and eventually scarring of the liver (cirrhosis) in about 3 to 4% of people with cystic fibrosis. Cirrhosis may increase the pressure in the veins entering the liver (portal hypertension), leading to enlarged, fragile veins at the lower end of the esophagus (esophageal varices), which can rupture and bleed profusely.
In almost all people with cystic fibrosis, the gallbladder is small, filled with thick bile, and does not function well. Some people develop gallstones, but only a small percentage develops symptoms. Surgical removal of the gallbladder is rarely needed.
People with cystic fibrosis often have impaired reproductive function. Almost all men have a low or absent sperm count (which makes them infertile) because one of the ducts of the testis (the vas deferens) has developed abnormally and blocks the passage of sperm. In women, cervical secretions are too thick, causing somewhat decreased fertility. However, many women with cystic fibrosis have carried pregnancies to term. The outcome of the pregnancy for both the mother and the newborn is often related to the mother's health status during pregnancy. Otherwise, sexual function is not impaired in men or women.
Other complications may include arthritis, chronic pain, problems sleeping and obstructive sleep apnea, kidney stones, kidney disease, depression and anxiety, sensorineural hearing loss and ear ringing (tinnitus) caused by exposure to medications that damage the ears (especially aminoglycosides), chronic sinus infections, and an increased risk of cancer of the bile ducts, pancreas, and intestines.
Diagnosis of Cystic Fibrosis
Newborn screening test
Sweat test
Genetic testing
Carrier screening
Other tests
Newborn screening
Screening tests for cystic fibrosis are done on all newborns in the United States and in other regions worldwide where such testing is available. A drop of blood is collected on a piece of filter paper and the level of trypsin (a digestive enzyme from the pancreas) is measured. If the trypsin level in the blood is high, some states in the US require repeat testing. In all cases, newborns who had a positive trypsin test undergo confirmatory testing, which includes sweat testing and genetic (DNA) testing. Most cases of cystic fibrosis are identified by newborn screening tests.
If newborn screening was not done, the diagnosis of cystic fibrosis is usually confirmed in infancy or early childhood, but cystic fibrosis occasionally goes undetected until adolescence or early adulthood.
Sweat test

A sweat test is done in newborns who have a positive newborn screening test and in infants, children, and adults who have symptoms that suggest cystic fibrosis. This test, which is done on an outpatient basis, measures the amount of salt in sweat. The drug pilocarpine is placed on the skin to stimulate sweating, and filter paper or thin tubing is placed against the skin to collect the sweat. The concentration of salt in the sweat is then measured. A salt concentration higher than normal confirms the diagnosis of cystic fibrosis in people who have symptoms of cystic fibrosis or who have a sibling with cystic fibrosis. Although this test can be done any time after a newborn is 48 hours old, collecting a large enough sweat sample from a newborn younger than about 2 weeks old may be difficult. A sweat test is done in newborns who have a positive newborn screening test and in infants, children, and adults who have symptoms that suggest cystic fibrosis. This test, which is done on an outpatient basis, measures the amount of salt in sweat. The drug pilocarpine is placed on the skin to stimulate sweating, and filter paper or thin tubing is placed against the skin to collect the sweat. The concentration of salt in the sweat is then measured. A salt concentration higher than normal confirms the diagnosis of cystic fibrosis in people who have symptoms of cystic fibrosis or who have a sibling with cystic fibrosis. Although this test can be done any time after a newborn is 48 hours old, collecting a large enough sweat sample from a newborn younger than about 2 weeks old may be difficult.
Genetic testing
Genetic testing for an abnormal CFTR gene can help diagnose cystic fibrosis in a newborn who has a positive newborn screening test result, in a person who shows one or more typical symptoms, or in a person who has a sibling with cystic fibrosis. Finding 2 abnormal cystic fibrosis genes (variants) is consistent with the diagnosis of cystic fibrosis. However, a positive sweat test is still needed to confirm the diagnosis. In addition, because typical genetic testing does not look for all of the more than 2,000 different cystic fibrosis variants, failure to detect 2 variants does not guarantee the person does not have cystic fibrosis (although the chance of having cystic fibrosis is very low). The disease can be diagnosed prenatally by doing genetic testing on the fetus using chorionic villus sampling or amniocentesis.
Some infants with a positive newborn screening test for cystic fibrosis can be hard to classify, even after sweat testing and genetic testing. These are infants who have no cystic fibrosis–related symptoms, sweat test results that are in-between the positive and negative ranges, and either no or only one cystic fibrosis gene variant. Doctors diagnose this group as having CFTR-related metabolic syndrome (CRMS), also called cystic fibrosis screen positive, inconclusive diagnosis (CFSPID). Although most of these infants remain healthy, later in life some (fewer than 10%) develop symptoms related to cystic fibrosis and are diagnosed with cystic fibrosis or a cystic fibrosis–related disorder. Thus, all of these children should be monitored regularly in a cystic fibrosis care center.
Some people, usually adults, develop symptoms that affect only one organ, often with an intermediate sweat test result and without 2 cystic fibrosis–causing variants. For example, symptoms may affect only the pancreas (resulting in pancreatitis), the lungs (resulting in bronchiectasis), or the male reproductive organs (resulting in infertility). They are diagnosed with a CFTR-related disorder.
Carrier testing
Carrier testing can be done for people who want to become parents or are looking for prenatal care. In particular, relatives of a person with cystic fibrosis may want to know whether they are at increased risk of having children with the disease, and they should be offered genetic testing and counseling. A blood sample is taken to help determine whether a person has a defective cystic fibrosis gene (variant).
Unless both potential parents have at least one such variant, their children will not have cystic fibrosis. If both parents carry a defective cystic fibrosis gene, each pregnancy has a 25% chance of producing a child with cystic fibrosis, a 50% chance of producing a child who is a carrier, and a 25% chance of producing a child with no defective cystic fibrosis genes.
Other tests
Because cystic fibrosis can affect several organs, other tests may be helpful. Pancreatic enzyme levels are usually reduced, and an analysis of the person’s stool may reveal low or undetectable levels of the digestive enzyme elastase (secreted by the pancreas) and high levels of fat. Even if pancreatic enzyme levels are normal initially, levels may change as cystic fibrosis progresses, so enzyme levels are measured periodically.
Blood tests are done to determine whether insulin secretion is reduced and blood sugar levels are high. Blood tests are also done to look for problems with the liver and to measure fat-soluble vitamin levels.
Doctors routinely take samples of material from the throat or coughed up from the lungs and culture it to identify bacteria in the airways and help them decide which antibiotics may be needed.
Pulmonary function tests may show that breathing is compromised and are good indicators of how well the lungs are functioning. These tests are generally done several times a year and whenever there is a decline in a person's health.
Chest x-rays and computed tomography (CT) of the chest may be helpful to document lung infection and the extent of lung damage. CT of the sinuses is done for people who have serious sinus symptoms, particularly if they have nasal polyps or are being considered for sinus surgery.
Treatment of Cystic Fibrosis
Routine immunizations
Antibiotics, inhaled medications to thin airway secretions, and airway clearance techniques to remove airway secretions
Medications that help prevent airways from narrowing (bronchodilators) and sometimes steroids (also called glucocorticoids or corticosteroids)
Pancreatic enzyme and vitamin supplements
High-calorie diet
In people with specific variants, CFTR modulators
A person with cystic fibrosis should have a comprehensive program of therapy directed by a doctor experienced with cystic fibrosis care—usually a pediatrician or an internist—along with a team of other doctors, nurses, a dietitian, a respiratory or physical therapist, and ideally a social worker, genetic counselor, pharmacist, and a mental health professional. The goals of therapy include long-term prevention and treatment of lung and digestive problems and other complications, maintenance of good nutrition, and encouragement of physical activity.
Children with cystic fibrosis may need mental health and social support because they may have challenges participating in normal childhood activities and consequently may feel isolated. Much of the burden of treating a child with cystic fibrosis falls on the parents, who should receive adequate information, training, and support so they can understand the disorder and the reasons for the treatments.
Adolescents need guidance and education as they transition to independence and assume responsibility for their care.
Adults need support as they deal with issues related to employment, relationships, health insurance, and deteriorating health.
Treatments for the lungs
The treatment of lung problems is focused on:
Preventing airway blockage
Controlling infection
The person should receive all routine immunizations, particularly for infections that can cause respiratory problems such as Haemophilus influenzae, influenza, measles, pertussis, pneumococcus, varicella, and COVID-19.
Airway clearance techniques, which include postural drainage, chest percussion, hand vibration over the chest wall, and encouragement of coughing (see Chest Physical Therapy), are started when cystic fibrosis is first diagnosed. Parents of a young child can learn these techniques and carry them out at home every day. Older children and adults can carry out airway clearance techniques independently by using special breathing devices, an inflatable vest that vibrates at a high frequency (a high-frequency oscillation vest), or special breathing maneuvers. Aerobic exercise, done regularly, can also help keep the airways clear.
Bronchodilators are medications that help prevent the airways from narrowing. People usually take bronchodilators by inhaling them.
Supplemental oxygen therapy may be needed in people with severe lung problems and a low level of oxygen in the blood.
A ventilator (breathing machine) generally does not help people with chronic respiratory failure. However, occasional, short periods of mechanical ventilation in the hospital may help during an acute infection, after a surgical procedure, or while waiting for a lung transplant. Some people are treated with a tight-fitting mask placed over the nose or nose and mouth. A mixture of oxygen and air is delivered under pressure through the mask. This technique, called bilevel positive airway pressure (BiPAP) or continuous positive airway pressure (CPAP), can help people maintain normal oxygen levels while they sleep.
Medications that help thin the thick mucus in the airways, such as dornase alfa or hypertonic saline (a highly concentrated salt solution), are widely used. These medications are inhaled through a nebulizer. They make it easier to cough up sputum, improve lung function, and may also decrease the frequency of serious respiratory tract infections. such as dornase alfa or hypertonic saline (a highly concentrated salt solution), are widely used. These medications are inhaled through a nebulizer. They make it easier to cough up sputum, improve lung function, and may also decrease the frequency of serious respiratory tract infections.
Steroids, (sometimes called glucocorticoids or corticosteroids) such as prednisone or dexamethasone, given by mouth can relieve symptoms in infants with severe bronchial inflammation, in people who have narrowed airways that cannot be opened with bronchodilators, and in people who have an allergic lung reaction to a type of fungus ((sometimes called glucocorticoids or corticosteroids) such as prednisone or dexamethasone, given by mouth can relieve symptoms in infants with severe bronchial inflammation, in people who have narrowed airways that cannot be opened with bronchodilators, and in people who have an allergic lung reaction to a type of fungus (allergic bronchopulmonary aspergillosis). Allergic bronchopulmonary aspergillosis is also treated with an antifungal medication given by mouth, by vein, or both.
Anti-inflammatory medications, such as a nonsteroidal anti-inflammatory drug (NSAID), or azithromycin, a macrolide antibiotic, are sometimes used to slow the deterioration of lung function.), or azithromycin, a macrolide antibiotic, are sometimes used to slow the deterioration of lung function.
Antibiotics
Antibiotics are often used to treat both acute and chronic respiratory tract infections in people with cystic fibrosis. A sample of coughed-up sputum or a swab of a sample from the back of the throat and tonsils is collected and tested at regular intervals during stable health, and also during periods of increasing symptoms, so that the infecting organism can be identified and the doctor can choose the medications most likely to control it. Staphylococcus aureus, including methicillin-resistant or methicillin-sensitive strains, and Pseudomonas species are commonly found. Many different antibiotics can be given by mouth to treat staphylococcal infections. To treat a new Pseudomonas infection, people are given the inhaled form of tobramycin, aztreonam, or colistin for 4 weeks.infection, people are given the inhaled form of tobramycin, aztreonam, or colistin for 4 weeks.
However, if the infection is severe, or results in significant worsening of symptoms or lung function, antibiotics given by vein (intravenously) may be needed. For this treatment, the aminoglycoside tobramycin (or sometimes amikacin) is combined with another antibiotic that specifically targets tobramycin (or sometimes amikacin) is combined with another antibiotic that specifically targetsPseudomonas. These other antibiotics include cephalosporins, penicillins, fluoroquinolones, and monobactams. This treatment often requires hospitalization, but part of the treatment may be given at home.
Taking the inhaled form of tobramycin or aztreonam every other month for a long time and also continuously taking an oral form of the antibiotic azithromycin 3 times each week may help control chronic Taking the inhaled form of tobramycin or aztreonam every other month for a long time and also continuously taking an oral form of the antibiotic azithromycin 3 times each week may help control chronicPseudomonas infection and slow the decline in lung function.
CFTR modulators
CFTR modulators are oral medications taken long term that improve the function of the defective protein made by variants in the CFTR gene.
There are several CFTR modulators or combinations for people who have specific variants These medications can be used to treat about 90% of people who have cystic fibrosis. Doctors give these medications to people based on their age and the variants they have.
IvacaftorIvacaftor can be given to people 1 month of age and older who have at least 1 copy of a specific cystic fibrosis variant.
The combination of lumacaftor and ivacaftor can be given to people 1 years of age and older who carry 2 copies of the F508del variant.
The combination of tezacaftor and ivacaftor can be given to people 6 years of age and older who carry 2 copies of the F508del variant or other specified variants.
The combination of elexacaftor, tezacaftor, and ivacaftor can be given to people 2 years of age and older who carry at least 1 copy of the F508del variant or 1 copy of certain rare variants.
The combination of vanzacaftor, tezacaftor, and ivacaftor can be given to people 6 years of age and older who carry at least 1 copy of the F508del variant or 1 copy of certain rare variants.
CFTR modulators can improve lung function, pancreas function, and quality of life; increase weight; and decrease the salt concentration in sweat and the frequency of lung infections and hospitalizations.
Doctors are working to develop medications that will help people with other variants that cause cystic fibrosis.
Enemas and stool softeners
Newborns who have blocked intestines may be treated with special enema solutions but often require surgery.
Older children and adults who have constipation or partially blocked intestines may be treated with stool softeners, enemas, and special solutions given by mouth or via a small, flexible plastic tube (nasogastric tube) that is passed through the nose or mouth into the stomach.
Diet and supplements
The diet should provide enough calories and protein for normal growth. Because digestion and absorption can be abnormal even when pancreatic enzyme supplements are used, most children need to consume 30 to 50% more calories than the usually recommended amount to ensure adequate growth. The proportion of fat should be normal to high. High-calorie oral supplements can provide extra calories for children and adults.
People who cannot absorb enough nutrients from food may need supplemental feedings given through a tube inserted into the stomach or small intestine.
People with cystic fibrosis should take double the usual recommended daily amount of fat-soluble vitamins (A, D, E, and K) in a special formulation that is more easily absorbed to prevent deficiencies of those vitamins.
Medications that stimulate the appetite may be helpful (although many people with cystic fibrosis have a strong appetite without any such medications). When people exercise, have a fever, or are exposed to hot weather, they should increase their fluid and salt intake.
Pancreatic enzyme supplements
People whose pancreas has been affected by cystic fibrosis must take capsules of pancreatic enzyme supplements with all meals and snacks. For infants, parents open the capsules and mix the contents with an acidic food such as applesauce so that the special coating on the pancreatic enzyme supplement does not dissolve before reaching the intestines. For some people, medications that reduce stomach acid, such as a histamine-2 blocker or a proton pump inhibitor, can improve the effectiveness of the pancreatic enzymes. Special milk formulas containing protein and fats that are easy to digest may help infants who have pancreatic problems and poor growth.
Insulin
People with cystic fibrosis who have diabetes need to take insulin injections. Oral medications for diabetes are not adequate treatment. need to take insulin injections. Oral medications for diabetes are not adequate treatment.
In addition to insulin, management includes nutrition counseling, a diabetes self-management program, and monitoring for eye and kidney complications. People also need special nutrition counseling because standard dietary recommendations for people with only diabetes or only cystic fibrosis are not adequate.
Surgery
At times, surgery may be needed to treat a collapsed lung, chronic sinus infections, severe chronic infection restricted to one area of the lung, bleeding from blood vessels in the esophagus, gallbladder disease, or obstruction of the intestine. Massive or recurring bleeding in the lung can be treated by a procedure called embolization, which blocks off the bleeding artery.
Liver transplantation has been successful for people who have bleeding resulting from esophageal varices or severe liver damage.
Double lung transplantation (that is, transplantation of both the right and left lungs) for severe lung disease may be necessary in some cases. For adults who have cystic fibrosis, average survival after a double lung transplant is about 9.5 years.
Other treatments
Medications to treat chronic inflammation of the sinuses (sinusitis) are needed because this problem is very common. Treatment options include flushing a saltwater solution through the nose (nasal saline irrigation), inhaling dornase alfa into the nose using a nebulizer, and irrigating the nose and sinuses with antibiotics. A steroid nasal spray is recommended to treat inflammation and swelling of the mucous membranes of the nose (allergic rhinitis).(sinusitis) are needed because this problem is very common. Treatment options include flushing a saltwater solution through the nose (nasal saline irrigation), inhaling dornase alfa into the nose using a nebulizer, and irrigating the nose and sinuses with antibiotics. A steroid nasal spray is recommended to treat inflammation and swelling of the mucous membranes of the nose (allergic rhinitis).
People who have heart failure are given medications (diuretics) to reduce the amount of fluid they retain. Diuretics help by increasing the amount of water the kidneys can get rid of from the body. People should also limit their intake of table salt and salty foods.
Injections of human growth hormone may improve lung function, increase height and weight, and reduce the rate of hospitalization. However, this medication is costly and inconvenient for people to receive, so doctors do not commonly prescribe it.
End-of-life issues
People who have cystic fibrosis and their family members need to have discussions with their doctor and care team about their prognosis and what types of treatment they want to receive. These discussions are especially important for people whose lung function is worsening. People need to be prepared for what is to come and know what treatments may be done to extend life. With advanced cystic fibrosis, people and their families need to discuss the potential benefits and burdens of lung transplantation.
Doctors should give people who have cystic fibrosis the information they need to make care choices. This includes guiding them through the complex process of understanding their prognosis and discussing end-of-life preferences and planning in a sensitive and timely manner. When aggressive treatments are no longer helpful, doctors may begin giving people treatments that aim only to relieve symptoms (called palliative care). People usually do best when they make such decisions for end-of-life care well in advance of needing to. Such early discussions are very important because, later on, the illness often prevents people from explaining their wishes. This process of making decisions in advance for end-of-life care is called advance care planning. Such planning should involve the execution of appropriate legal documents that reflect the person's wishes regarding end-of-life care.
Prognosis for Cystic Fibrosis
The severity of cystic fibrosis varies greatly from person to person regardless of age. The severity is determined largely by how much the lungs are affected. In the United States, people with cystic fibrosis who were born in the year 2023 are predicted to live to about 68 years of age. The outlook for longer survival has improved steadily as people are diagnosed earlier and treatments can now postpone some of the changes that occur in the lungs. Long-term survival is significantly better in people who do not develop pancreatic problems.
However, deterioration is inevitable, leading to loss of lung function and eventually death. People with cystic fibrosis usually die of respiratory failure after many years of deteriorating lung function. A small number, however, die of heart failure, liver disease, bleeding into the airways, or complications of surgery including lung and/or liver transplantation. Despite their many problems, people with cystic fibrosis may lead productive lives and often attend school or work until shortly before death.
More Information
The following English-language resource may be useful. Please note that The Manual is not responsible for the content of this resource.
Drug Information for the Topic


