





There are several different birth defects that affect the kidneys (the organs that filter waste from the blood to make urine). These defects are not usually apparent at the doctor's examination and require tests to evaluate the urinary tract.
(See also Overview of Urinary Tract Birth Defects.)
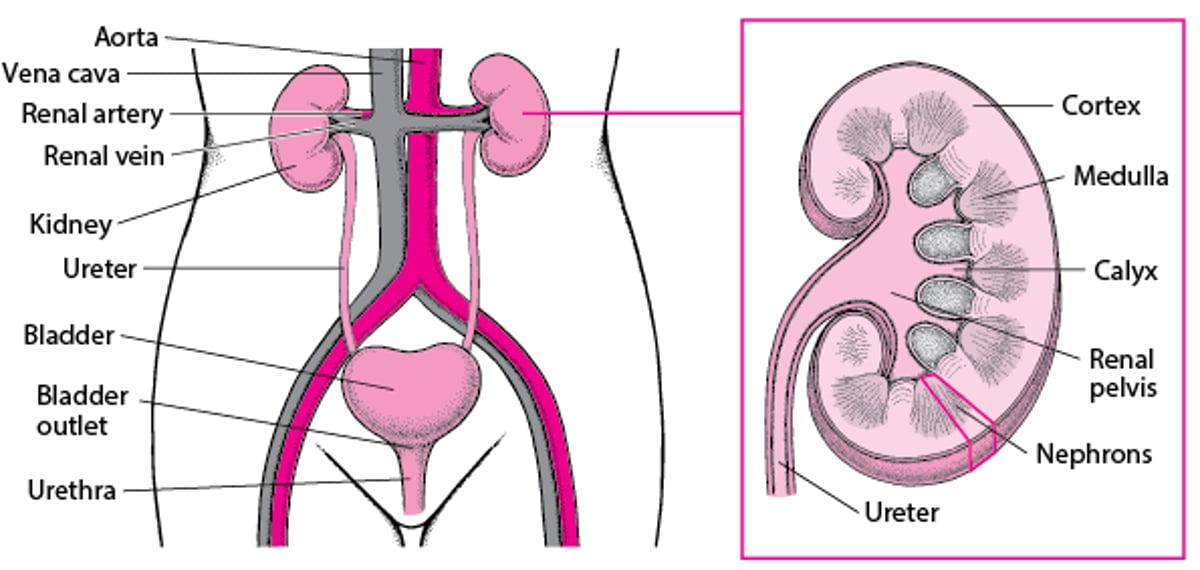
A Look Inside the Urinary Tract

Complications of birth defects of the kidneys
There are many types of birth defects of the kidneys. Many of these defects
Block or slow the flow of urine out of the kidneys
Blockage of urine flow can cause urine to become stagnant and result in urinary tract infections (UTIs) or formation of kidney stones. Blockage of urine flow also can raise the pressure inside the kidneys and damage them over time. Kidney damage can cause high blood pressure and, rarely, kidney failure.

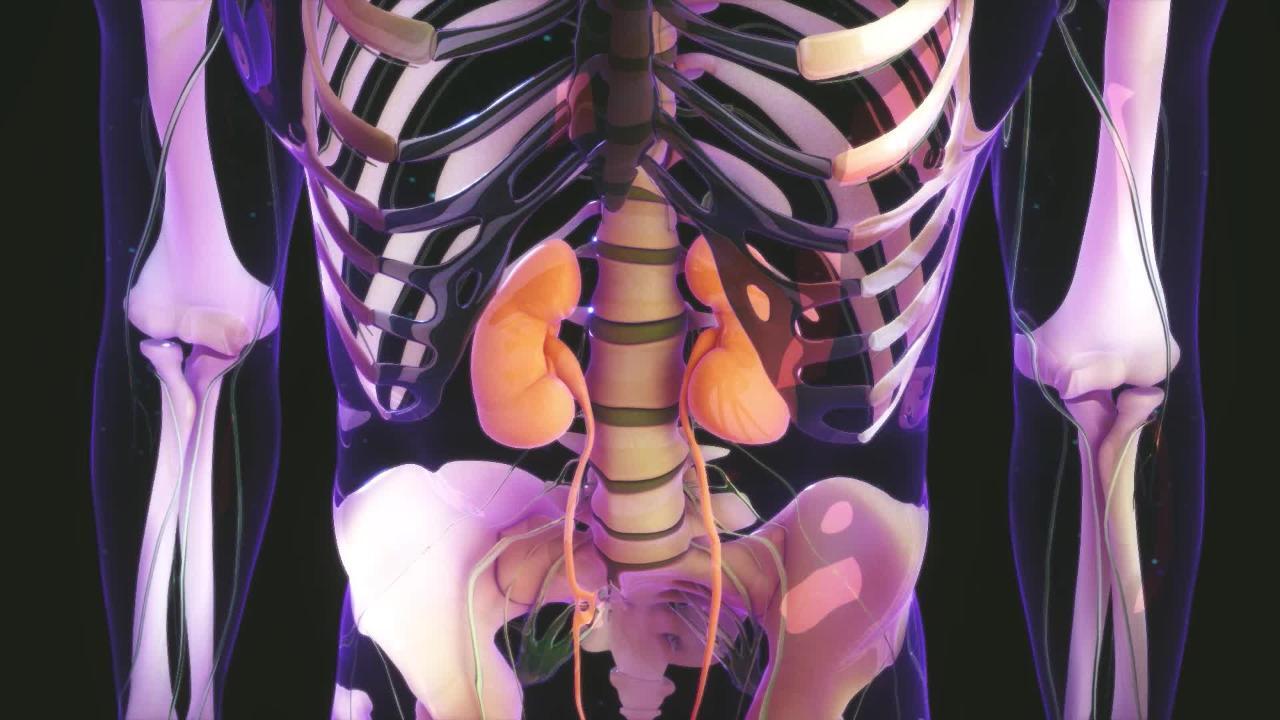
Types of Kidney Defects
A number of birth defects may result in abnormal kidneys. Kidneys may be
In the wrong location (ectopic kidneys)
Rotated the wrong way (malrotation)
Joined together (horseshoe kidney or fused kidneys)
Missing (agenesis)
Poorly functioning
Containing fluid-filled sacs (cysts), such as in polycystic kidney disease and multicystic dysplastic kidney
Kidneys in the wrong location and position
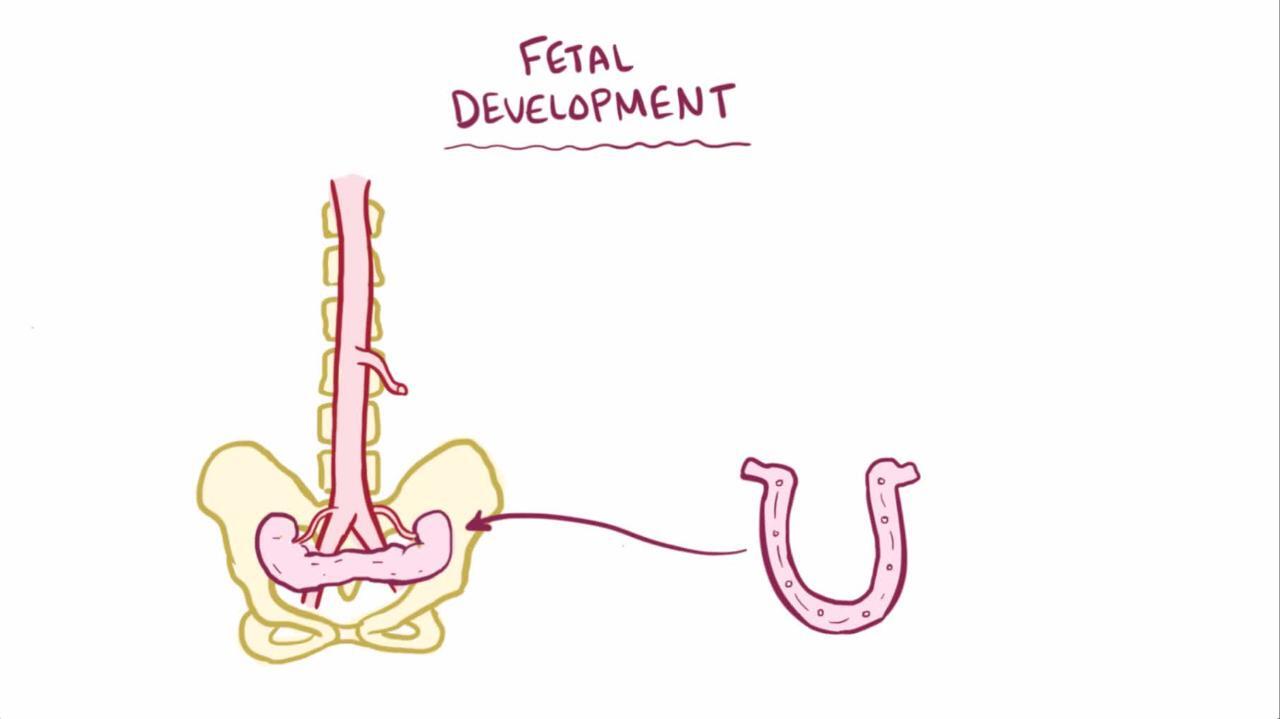
In the fetus, kidneys first develop in the pelvis and then move up and rotate into their normal location in the upper abdomen. If the kidneys are in the wrong place (ectopic kidneys) or are not rotated properly (malrotation), the normal drainage of urine from the kidneys through the ureters (the tubes that transport urine from the kidneys to the bladder) to the bladder (the expandable, muscular sac that holds urine) may be blocked.
Children who have blockage that causes frequent urinary tract infections or other problems may need surgery. However, many children do not have symptoms but may still need surgery.
Horseshoe kidneys
A horseshoe kidney is the most common fused kidney defect. In horseshoe kidney, the fetus's 2 kidneys join together (fused) into a single, horseshoe-looking shape. Because they are joined together, the kidneys do not usually move up and rotate into their normal location and also may not develop properly. Because of these factors, a horseshoe kidney may not drain properly, increasing the risk of urinary tract infections, kidney stones, and kidney damage. However, over half of children with a horseshoe kidney never have any symptoms. Children who have a horseshoe kidney may also have other birth defects.

Missing kidneys
In some children, 1 or both of the kidneys may not develop at all (kidney agenesis). Children who are missing both kidneys cannot survive.
In children who are missing only 1 kidney, the remaining kidney usually develops normally and often becomes bigger than normal to compensate for the missing one. In such cases, the child is expected to have a normal life expectancy and does not need any treatment.
Poorly functioning kidneys
Sometimes a kidney does not form properly and may not function properly or at all (kidney dysplasia). When only 1 kidney functions properly, it may become bigger than normal to make up for the loss of function of the other kidney. Because all of the functions normally done by 2 kidneys can be carried out adequately by 1 healthy kidney, children who have only 1 functioning kidney often lead normal, healthy lives. However, if dysplasia is widespread and affects both kidneys, children may need treatment to replace the kidney function. That is, they will need a kidney transplant or dialysis (a way to remove waste products and excess fluids from the body).
Polycystic kidney disease
Polycystic kidney disease is a hereditary disorder in which many fluid-filled sacs (cysts) form in both kidneys. The kidneys grow larger but have less functioning tissue.
There are several forms of polycystic kidney disease. One form, autosomal dominant polycystic kidney disease, usually does not appear until adulthood and usually causes mild symptoms.
Autosomal recessive polycystic kidney disease is the rare form of this disorder that begins during childhood. In this disorder, the cysts become very large and cause serious illness. A severely affected newborn may die shortly after birth because kidney failure can develop before birth, leading to poor development of the lungs. The liver is also affected, and a child with this disorder tends to develop portal hypertension, or high pressure in the blood vessels that connect the intestine and the liver (portal system). Eventually, liver failure and chronic kidney disease occur. Infants who survive the newborn period may need kidney (and sometimes liver) transplantation.
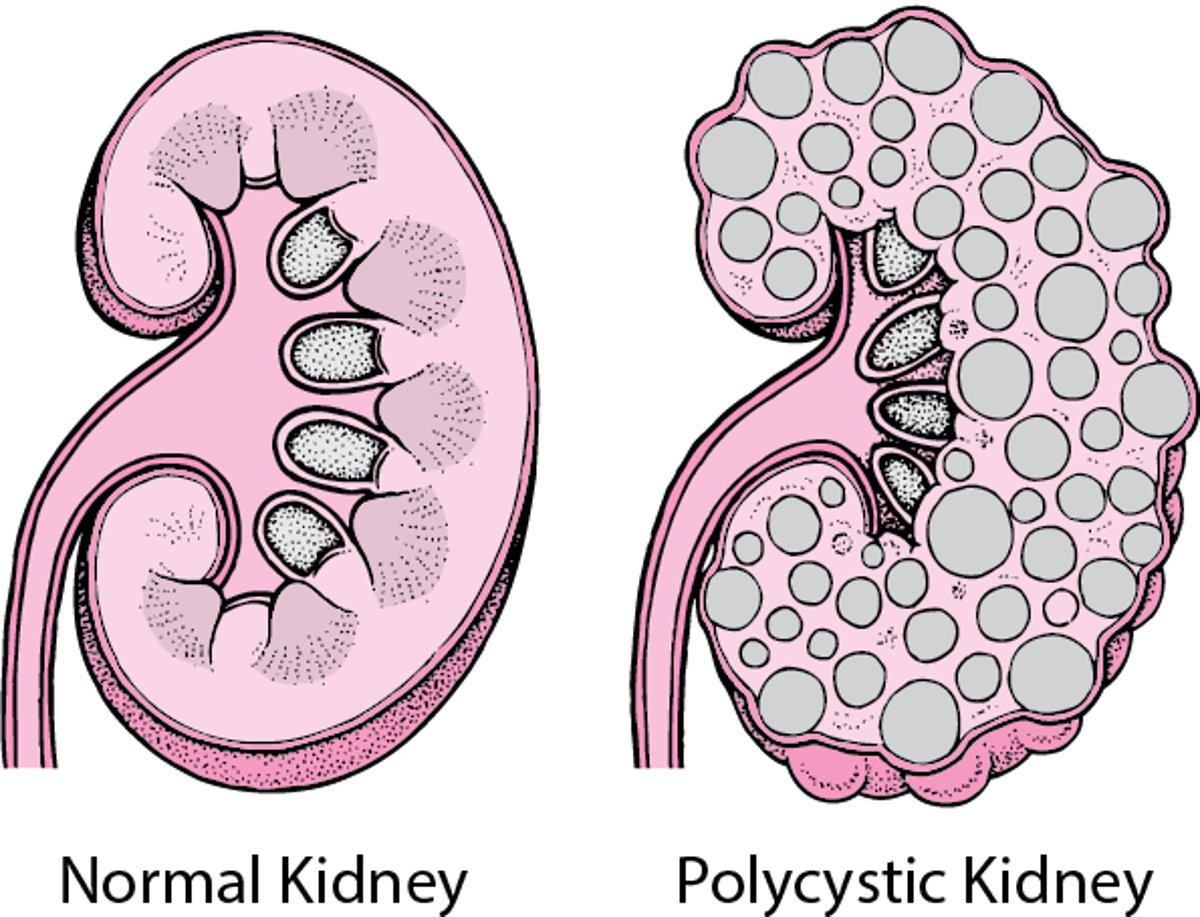
Polycystic Kidney Disease
In polycystic kidney disease, many cysts form in both kidneys. The cysts gradually enlarge, destroying some or most of the normal tissue in the kidneys. |

Multicystic dysplastic kidney
Multicystic dysplastic kidney (MCDK) is the most common cyst-causing malformation of the kidneys in children. In MCDK, the kidney does not develop normally and instead has multiple fluid-filled sacs (cysts) that grow inside the kidney and take over the normal kidney tissue. The kidney does not function at all.
MCDK typically affects only 1 kidney. If it affects both kidneys, the fetus does not survive. However, children who have only 1 affected kidney often have a good outlook, depending on what other abnormalities, if any, they have. Sometimes the unaffected kidney has other defects that can cause blockage of urine flow or reflux of urine.
MCDK is usually detected during routine prenatal ultrasound, and most children have periodic ultrasounds after birth to determine whether the unaffected kidney is functioning properly. The affected kidney almost always shrinks and disappears. If the kidney does not go away, doctors sometimes remove it later in childhood.
Because the unaffected kidney usually becomes bigger over time to compensate for the affected one, children usually have normal kidney function. If children have urinary blockage that causes frequent urinary tract infections or other problems in the functioning kidney, they may need surgery.
Diagnosis of Kidney Birth Defects
Prenatal ultrasound and tests before birth, and imaging tests and physical examinations after birth
Sometimes biopsy of the kidney
Sometimes genetic testing
Before birth, urinary tract defects are often discovered by doctors during routine prenatal ultrasound or other routine screening tests for hereditary disorders.
After birth, kidney defects that cause no symptoms are often discovered when imaging studies are done for other reasons. If doctors suspect a child has a kidney defect, they typically do imaging tests such as ultrasounds, computed tomography (CT), nuclear scans, or magnetic resonance imaging (MRI). Rarely, doctors do intravenous urography.
Children who have dysplasia may undergo a biopsy of the kidney (a sample of kidney tissue is removed and examined under a microscope).
Because polycystic kidney disease is inherited, people who have it may have genetic testing done to help them understand whether they might pass on to their children the defective gene that causes the disease.
Treatment of Kidney Birth Defects
Sometimes surgical procedures
Sometimes a kidney transplant or dialysis
Treatment of kidney birth defects depends on the specific defect but, in general,
Children who have blockage of urine flow that causes frequent urinary tract infections, pain, damage to the kidney, or other problems may need surgery.
Children whose kidney function is poor may need a kidney transplant or dialysis.





