





Marfan syndrome is a rare hereditary disorder of connective tissue that causes abnormalities of the eyes, bones, heart, and blood vessels.
This syndrome is caused by mutations in the gene that codes for a protein called fibrillin.
Typical symptoms can range from mild to severe and include long arms and fingers, flexible joints, and heart and lung problems.
The diagnosis is based on symptoms and genetic testing.
There is no cure for Marfan syndrome or any way to correct the abnormalities in the connective tissue, but complications can be treated with medications and surgery.

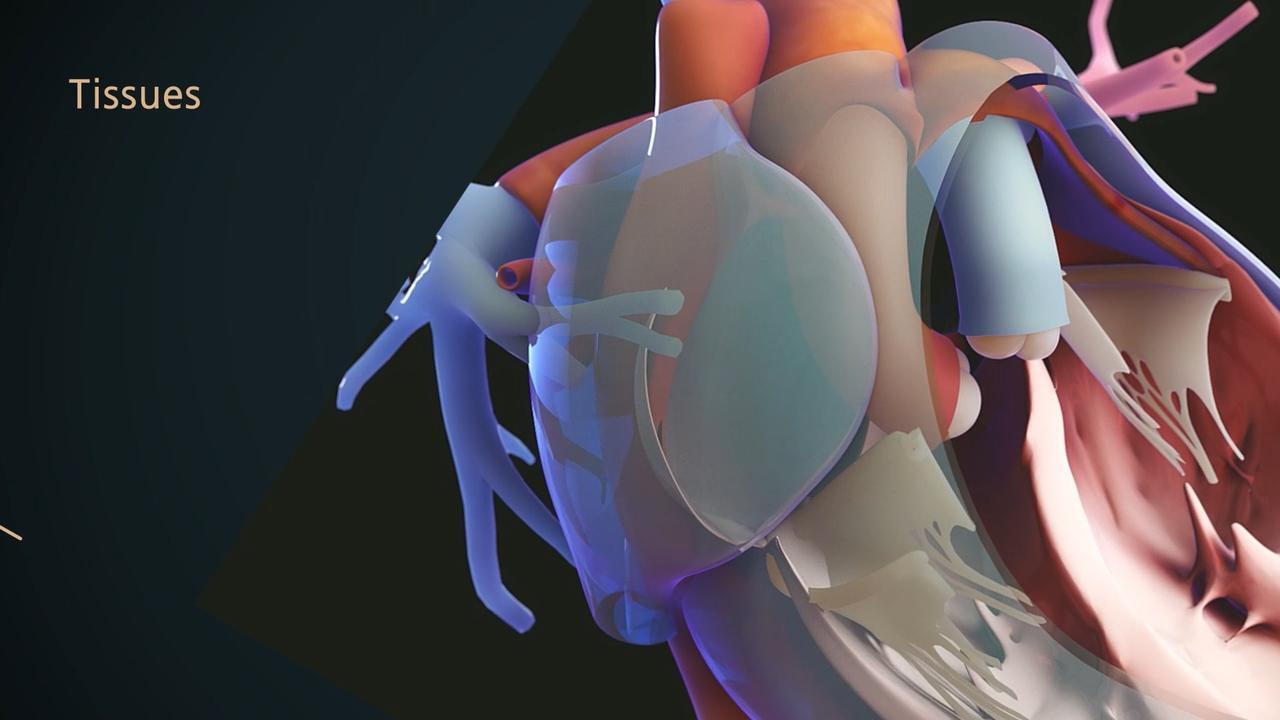
Marfan syndrome is caused by mutations in the FBN1 gene. This gene codes for a protein called fibrillin. Fibrillin helps connective tissue maintain its strength. Connective tissue is the tough, often fibrous tissue that binds the body's structures together and provides support and elasticity.
If the FBN1 gene is mutated, some fibers and other parts of connective tissue undergo changes that ultimately weaken the tissue. The weakening affects bones and joints as well as internal structures, such as the heart, blood vessels, and eyes and sometimes the lungs, brain, and spinal cord. Weakened tissues stretch, distort, and can even tear. For example, the aorta (the main artery of the body) may weaken, bulge, or tear. Weak tissues in a heart valve can cause the valve to leak. Connective tissues that join structures may weaken or break, separating formerly attached structures. For example, the lens or retina of the eye may separate from its normal attachments.
Symptoms of Marfan Syndrome
There are many different manifestations of Marfan syndrome, but doctors typically suspect it in people who have a combination of long limbs, problems with the aorta, and lenses of the eyes that may be in the wrong place.
Symptoms of Marfan syndrome can range from mild to severe. Many people with Marfan syndrome never notice symptoms. In some people, symptoms may not become apparent until adulthood. Some people who have a very severe form of Marfan syndrome called neonatal Marfan syndrome begin showing symptoms when they are young infants (or even before birth, when the symptoms are seen by doctors on an ultrasound).
Heart problems
The most dangerous complications develop in the aorta, which is the large blood vessel that carries blood from the heart to the body. Weakness may develop in the connective tissue of the wall of the aorta. The weakened wall may result in blood leaking between the inner layers of the aorta’s wall (aortic dissection), which causes a tear, or in a bulge (aortic aneurysm) that can rupture. These problems sometimes develop before a child is 10 years old.
Pregnancy increases the risk of aortic dissection in people who have an enlarged aorta. Depending on the degree of enlargement, a caesarean delivery (C-section) may be recommended to minimize the risk.
If the aorta gradually widens or dilates, the aortic valve, which leads from the heart into the aorta, may begin to leak (called aortic regurgitation). The mitral valve, which is located between the left atrium and ventricle, may leak (mitral regurgitation) or bulge backward into the left atrium (mitral valve prolapse).
These heart valve abnormalities can impair the heart’s ability to pump blood.
Serious infections (infective endocarditis) can develop in abnormal heart valves.
Musculoskeletal problems

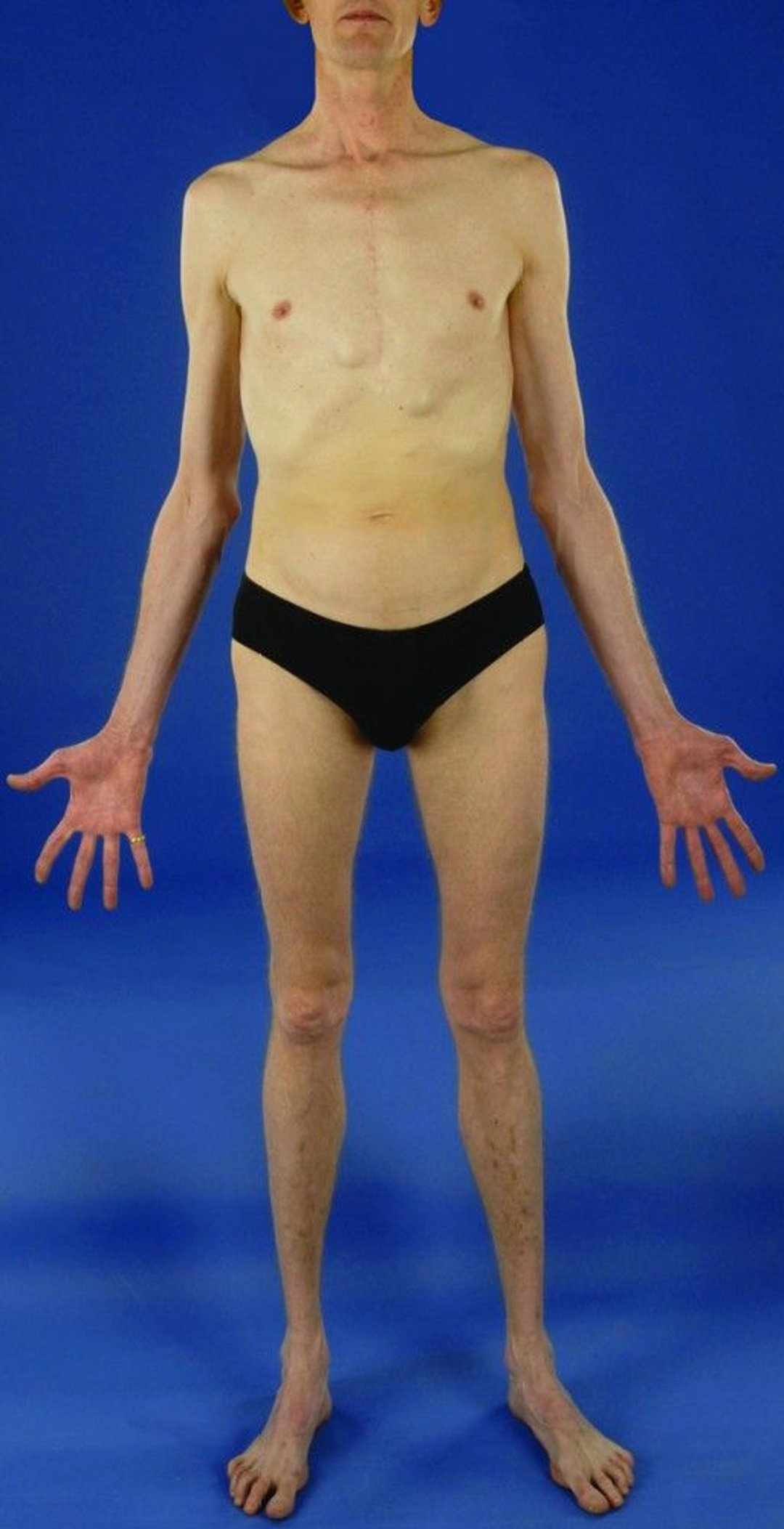
This man who has Marfan syndrome is unusually tall. His arm span exceeds his height. He has a scar down the middle of his chest because he has also undergone replacement of the aortic valve and the part of the aorta (the large blood vessel that carries blood from the heart to the body) that is closest to the heart.
© Springer Science+Business Media

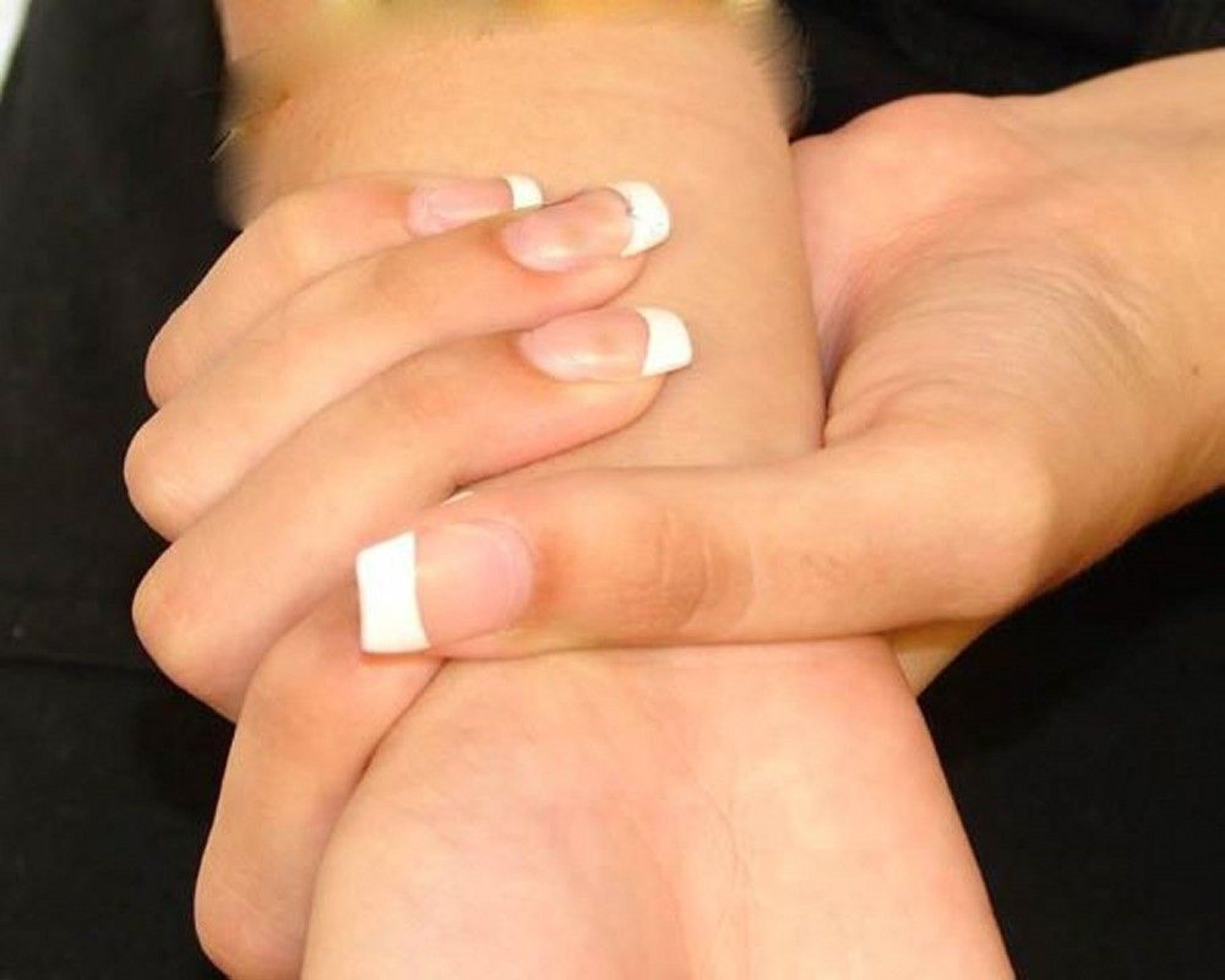
Marfan syndrome is characterized by abnormally long fingers. In this photo, the woman's thumb overlaps her index finger when her hand is wrapped around her wrist.
Photo courtesy of David D. Sherry, MD.
People with Marfan syndrome are taller than expected for their age and in comparison to family members. Their arm span (the distance between fingertips when the arms are outstretched) is greater than their height. Their fingers are long and thin. Often, the breastbone (sternum) is deformed and is pushed outward (called pectus carinatum) or inward (called pectus excavatum). The joints may be very flexible. Flat feet, a deformity of the knee joint that causes the knee to bend backward, and a humpback with an abnormal curve of the spine (kyphoscoliosis) are common, as are hernias. Usually, the person has little fat under the skin.
People also may have a skull that is long and narrow, eyes that are sunken and slant downward at the outer corners, a lower jaw that is set further back and causes an overbite, and cheekbones that are underdeveloped. The roof of the mouth often has a high arch.
Eye problems
The lens of one or both eyes may not be in the correct position (dislocated). People are very nearsighted (unable to see distant objects clearly). The light-sensitive area at the back of the eye (retina) may detach suddenly from the rest of the eye (see Detachment of the Retina).
Dislocation of the lens and detachment of the retina may cause permanent loss of vision.
Lung problems
Air-filled sacs (cysts) may develop in the lungs. The cysts may rupture, bringing air into the space that surrounds the lungs (pneumothorax). These disorders can cause chest pain and shortness of breath.
Brain and spinal cord problems
The sac that surrounds the spinal cord may widen (called dural ectasia). Dural ectasia is common among people with Marfan syndrome and most frequently occurs in the lower portions of the spine. It may cause headache, lower back pain, or other neurologic problems such as bowel or bladder weakness.
Diagnosis of Marfan Syndrome
A doctor's evaluation
Genetic testing
Echocardiography of the heart
Magnetic resonance imaging (MRI) of the heart and spine
X-rays of the spine and pelvis
Eye examinations
Doctors may suspect Marfan syndrome if an unusually tall, thin person has any of the characteristic symptoms or if Marfan syndrome has been diagnosed in other family members (first-degree relatives such as the father, mother, or a sibling). Doctors also base the diagnosis on specific criteria regarding the extent to which certain organ systems, such as the heart, eyes, and bones, are affected.
Doctors do genetic blood tests to analyze genes and help diagnose Marfan syndrome.
As part of the testing, doctors do several imaging tests, such as echocardiography of the heart and aorta; MRI of the heart and brain; and x-rays of the spine and pelvis. They also do eye examinations.
Treatment of Marfan Syndrome
Beta-blockers, angiotensin II receptor blockers, or both
Sometimes surgical repair of the aorta and valves, lenses of the eye, or retina
Sometimes a brace and sometimes surgical repair for curving of the spine
Sports and exercise guidelines
There is no cure for Marfan syndrome or any way to correct the abnormalities in the connective tissue, but treatment of Marfan syndrome is aimed at preventing and fixing dangerous complications.
People should have their heart, bones, and eyes checked every year to see if they are getting worse.
People should receive genetic counseling. People and their families may obtain additional information from The Marfan Foundation.
Medications
Doctors give beta-blockers (such as atenolol and propranolol), angiotensin II receptor blockers (such as losartan and irbesartan), or a combination of the two to all adults with Marfan syndrome. Medications are given to children depending on their age and on the size of their aorta. Beta-blockers slow the heart rate and decrease the force of heart contractions. Angiotensin II receptor blockers lower the blood pressure. These medications are given to make blood flow more gently through the aorta.
Surgery
However, if the aorta has widened, doctors may offer people the option to surgically repair or replace the affected section before problems start. Severe valve regurgitation is also surgically repaired. Pregnant people with Marfan syndrome are at especially high risk of complications with their aorta, so doctors may discuss with them repair of the aorta before they become pregnant.
Doctors are able to remove and sometimes replace dislocated lenses, repair detached retinas, and correct vision.
A brace is used to treat abnormal curving of the spine (scoliosis) for as long as possible. However, some children need a surgical procedure to correct the curve if it is severe.
Physical activity
Some general guidelines about physical activity for people with Marfan syndrome are to avoid competitive and contact sports that put added stress on the aorta, cause eye injuries, could possibly damage loose ligaments and joints, and increase the risk of collision or bodily contact. However, physical activity is beneficial for all people, including those with Marfan syndrome. Health care professionals can help people determine risks and help them make decisions about physical activity.
Prognosis for Marfan Syndrome
The expected life span for people who have Marfan syndrome is nearly the same as that for people who do not have this disorder. However, quality of life is decreased because people with Marfan syndrome have chronic pain and physical limitations and are often affected mentally by the disorder.
More Information
The following English-language resource may be useful. Please note that The Manual is not responsible for the content of this resource.
Drug Information for the Topic





