

Many factors are involved in causing and permitting the unregulated proliferation of cells that occurs in cancer.
(See also Overview of Cancer.)
Cellular Kinetics
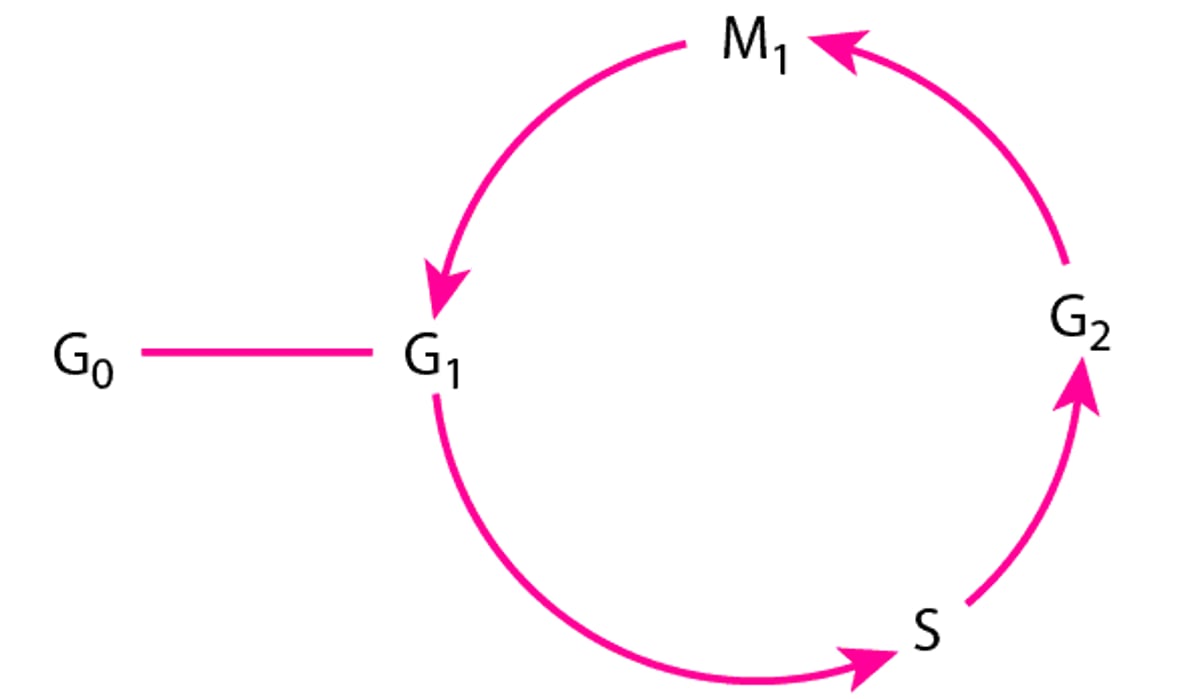
Generation time is the time required for a cell to complete a cycle in cell division (see figure ) and give rise to 2 daughter cells. Cancer cells, particularly those arising from the bone marrow or lymphatic system, may have a short generation time, and there usually are a smaller percentage of cells in G0 (resting phase). Initial exponential tumor growth is followed by a plateau phase when cell death nearly equals the rate of formation of daughter cells. The slowing in growth rate may be related to exhaustion of the supply of nutrients and oxygen for the rapidly expanding tumor. Small tumors have a greater percentage of actively dividing cells than do large tumors.
A subpopulation of cells within a cancer has the properties of stem cells. Thus, these cells are capable of entering a proliferative state. They are also less susceptible to injury by chemotherapy agents or irradiation. They are believed to repopulate cancers after chemotherapy and/or radiation treatment.
The cellular kinetics of particular cancers is an important consideration in the design of antineoplastic medication regimens and may influence the dosing schedules and timing intervals of treatment. Many antineoplastic medications, such as antimetabolites, are most effective when cells are actively dividing. Some medications work only during a specific phase of the cell cycle, requiring prolonged administration to catch dividing cells during the phase of maximal sensitivity.
The Cell Cycle
G0 = resting phase (nonproliferation of cells); G1 = variable pre-DNA synthetic phase (12 h to a few days); S = DNA synthesis (usually 2 to 4 h); G2 = post-DNA synthesis (2 to 4 h)—a tetraploid quantity of DNA is found within cells; M1 = mitosis (1 to 2 h). |

Cancer Growth and Metastasis
As a cancer grows, nutrients are provided by direct diffusion from the circulation. Local growth is facilitated by enzymes (eg, proteases) that destroy adjacent tissues. As the cancer volume increases, the cancer may release angiogenesis factors, such as vascular endothelial growth factor (VEGF), which promote formation of new blood vessels required for further growth.
A cancer may release cells into the blood and lymphatics at a very early stage of development. From animal models, it is estimated that a 1-cm tumor sheds > 1 million cells/24 hours into the blood. Circulating cancer cells are present in many patients with advanced cancer and even in some with localized disease. Although most circulating cancer cells die, an occasional cell may penetrate into tissues, generating a metastasis at a distant site. Metastases grow in much the same manner as the primary cancer and may subsequently give rise to other metastases. Most patients with cancer die from metastases rather than the cancer at the primary site.
Experiments suggest that the abilities to invade, migrate, and successfully implant and stimulate new blood vessel growth are all important properties of the cells that cause metastases, which are likely a subpopulation of the primary cancer.
The Immune System and Cancer
Cancer cells often present neoantigens on their cell surface that can be detected as "non-self" by the immune system, resulting in an attack by the immune system. When and if this immune attack is effective, a cancer may never develop. Destruction of cancer cells may be complete, in which case the cancer never appears. However, some cancers have or acquire the ability to avoid detection and/or destruction by the immune system, allowing them to proliferate.
It is not clear why people with a congenital or acquired immune deficiency have an increased risk of only some uncommon cancers such as melanoma, renal cell carcinoma, and lymphomas and not of more common cancers of the lung, breast, prostate, and colon. Most cancers in which the immune system is effective are caused by viruses.
Under selective (eg, evolutionary) pressure, cancer cells can express checkpoint proteins. Checkpoint proteins are cell-surface molecules, which signal T cells that the cells expressing them are normal and should not be attacked. An example is the programmed death ligand 1 (PD-L1) protein, which is recognized by the PD-1 molecule on T cells; when PD-L1 binds to PD-1 on a T cell, an immune attack is prevented. Cancer therapy using monoclonal antibodies termed checkpoint inhibitors, which block PD-L1 or PD-1, allow the immune system to attack protected cancer cells. Cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) is another checkpoint protein that prevents immune system attack and can be similarly blocked by a specific antibody. Because checkpoint proteins can be present on normal cells, checkpoint inhibitor therapy may also induce an autoimmune response.
Genetically engineered T cells (referred to as chimeric antigen receptor T-cell [CAR-T] therapy) can also be used in immune therapy. In this process, T cells are removed from a patient and genetically modified to express receptors containing a recognition domain for a specific antigen coupled to intracellular signaling domains that activate the T cell. When the modified T cells are infused, they can attack cells bearing the target antigen. Usually, the target antigen is lineage-specific, not cancer-specific. CAR-T-cell therapy is most effective against B-cell cancers such as B-cell acute lymphoblastic leukemia, B-cell lymphomas, and plasma cell myeloma (multiple myeloma). The efficacy of CAR-T-cell therapy against common solid cancers is not yet established.
Molecular Abnormalities in Cancer
Genetic mutations are responsible for the generation of cancer cells and are thus present in all cancers. These mutations alter the quantity or function of protein products that regulate cell growth and division and DNA repair. Two major categories of mutated genes are
Oncogenes
Tumor suppressor genes
Oncogenes
Oncogenes are abnormal forms of normal genes (proto-oncogenes) that regulate various aspects of cell growth and differentiation. Mutations in these genes may result in direct and continuous stimulation of the pathways (eg, cell surface growth factor receptors, intracellular signal transduction pathways, transcription factors, secreted growth factors) that control cellular growth and division, cellular metabolism, DNA repair, angiogenesis, and other physiologic processes.
There are > 100 known oncogenes that may contribute to human neoplastic transformation. For example, the RAS gene encodes the ras protein, which carries signals from membrane-bound receptors down the RAS-MAPKinase pathway to the cell nucleus, and thereby regulates cell division. Mutations may result in the inappropriate activation of the ras protein, leading to uncontrolled cell growth. The ras protein is abnormal in about 25% of human cancers (1).
Other oncogenes have been implicated in specific cancers. These include
HER2 (amplified in breast and gastric cancer and less commonly in lung cancer)
BCR::ABL1 (a chimeric gene present in chronic myeloid leukemia and some B-cell acute lymphocytic leukemias)
CMYC (Burkitt lymphoma)
NMYC (small cell lung cancer, neuroblastoma)
EGFR (adenocarcinoma of the lung)
EML4ALK (a chimeric gene present in adenocarcinoma of the lung)
KRAS (pancreatic cancer, lung cancer)
Specific oncogenes may have important implications for diagnosis, therapy, and prognosis (see individual discussions under the specific cancer type).
Oncogenes typically result from
Acquired somatic cell point mutations (eg, from chemical carcinogens)
Gene amplification (eg, an increase in the number of copies of a normal gene)
Translocations (in which pieces of different genes are joined to form a unique sequence)
These changes may either increase the activity of the gene product (protein) or change its function. Occasionally, mutation of genes in germ cells results in inheritance of a cancer predisposition.
Tumor suppressor genes
Genes such as TP53, BRCA1, and BRCA2 play a role in normal cell division and DNA repair and are critical for detecting inappropriate growth signals or DNA damage in cells. If these genes, as a result of inherited or acquired mutations, become unable to function, the system for monitoring DNA integration becomes inefficient, cells with spontaneous genetic mutations persist and proliferate, and tumors result.
As with most genes, 2 alleles are present that encode for each tumor suppressor gene. A defective copy of one gene may be inherited, leaving only one functional allele for the individual tumor suppressor gene. If a mutation is acquired in the functional allele, the normal protective mechanism of the second normal tumor suppressor gene is lost.
The important regulatory protein, p53, prevents replication of damaged DNA in normal cells and promotes cell death (apoptosis) in cells with abnormal DNA. Inactive or altered p53 allows cells with abnormal DNA to survive and divide. TP53 mutations are passed to daughter cells, conferring a high probability of replicating error-prone DNA, and neoplastic transformation results. TP53 is defective in many human cancers.
BRCA1 and BRCA2 mutations that decrease function increase risk of breast and ovarian cancers.
Another example, the retinoblastoma (RB) gene encodes for the protein Rb, which regulates the cell cycle by stopping DNA replication. Mutations in the RB gene family occur in many human cancers, allowing affected cells to divide continuously.
As with oncogenes, mutation of tumor suppressor genes such as TP53 or RB in germ cell lines may result in vertical transmission and a higher incidence of cancer in offspring.
Chromosome abnormalities
Chromosome abnormalities can occur through deletion, translocation, duplication, and other mechanisms. If these alterations activate or inactivate genes that result in a proliferative advantage over normal cells, then a cancer may develop. Chromosome abnormalities occur in many human cancers. In some congenital diseases (Bloom syndrome, Fanconi anemia, Down syndrome), DNA repair processes are defective and chromosome breaks are frequent, putting children at high risk of developing acute leukemia and lymphomas.
Other influences
Most epithelial cancers likely result from a sequence of mutations that lead to neoplastic conversion. For example, the development of colon cancer in familial polyposis takes place through a sequence of genetic events: epithelium hyper-proliferation (loss of a suppressor gene on chromosome 5), early adenoma (change in DNA methylation), intermediate adenoma (overactivity of the RAS oncogene), late adenoma (loss of a suppressor gene on chromosome 18), and finally, cancer (loss of a gene on chromosome 17). Further genetic changes may be required for metastasis.
Telomeres are nucleoprotein complexes that cap the ends of chromosomes and maintain their integrity. In normal tissue, telomere shortening (which occurs with aging) results in a finite limit in cell division. The enzyme telomerase, if activated in tumor cells, provides for new telomere synthesis and allows continuous proliferation of cancer cells. Inherited abnormalities in the genes responsible for telomere regeneration result in shortened telomeres and an increased risk of skin, gastrointestinal, and bone marrow cancers.
Molecular abnormalities reference
1. Keeton AB, Salter EA, Piazza GA. The RAS-Effector Interaction as a Drug Target. Cancer Res 2017;77(2):221-226. doi:10.1158/0008-5472.CAN-16-0938
Environmental Factors and Cancer
Infections
Viruses contribute to the pathogenesis of some human cancers (see table Cancer-Associated Viruses). Pathogenesis may occur through the integration of viral genetic elements into host DNA (1). These new genes are expressed by the host; they may affect cell growth or division or disrupt normal host genes required for control of cell growth and division. Alternatively, viral infection may result in immune dysfunction, leading to decreased immune surveillance for early tumors. HIV infection increases the risk of several cancers (see Cancers Common in Patients With HIV Infection).
Cancer-Associated Viruses
Virus | Some Associated Cancers |
|---|---|
Epstein-Barr virus | |
Hepatitis-B and -C viruses | |
Human papillomaviruses | Tonsil cancer and sometimes other head and neck cancers |
Human T-lymphotropic virus-1 (HTVL-1) | |
Kaposi sarcoma-associated herpesvirus | Multicentric Castleman disease Primary effusion lymphoma |
Merkel cell polyomavirus |
Bacteria can also cause cancer. Helicobacter pylori infection increases the risk of several kinds of cancer (gastric adenocarcinoma, gastric lymphoma, mucosa-associated lymphoid tissue [MALT] lymphoma) (2, 3).
Parasites of some types can lead to cancer. Schistosoma haematobium and Clonorchis sinensis cause chronic inflammation and fibrosis of the bladder, which may lead to cancer. Opisthorchis sinensis has been linked to carcinoma of the pancreas and bile ducts (4).
Radiation
Ultraviolet radiation can induce skin cancer (eg, basal and squamous cell carcinoma, melanoma) by damaging DNA. This DNA damage consists of formation of thymidine dimers, which may escape excision and re-synthesis of a normal DNA strand. Patients with inherent defects in DNA repair (eg, xeroderma pigmentosum) or immune suppression by medications or an underlying disease are particularly prone to skin cancers from ultraviolet exposure.
Ionizing radiation is carcinogenic. For example, survivors of the atomic bomb explosions in Hiroshima and Nagasaki have a higher-than-expected incidence of leukemias and solid cancers. Similarly, exposure to therapeutic radiation may lead to leukemia, breast cancer, sarcomas, and other solid cancers years after exposure. Exposure to X-rays for diagnostic imaging studies is thought to increase risk of cancer (see Risks of Medical Radiation). Occupational exposure (eg, to uranium by mine workers) is linked to development of lung cancer, particularly in people who smoke. Long-term exposure to occupational irradiation or to internally deposited thorium dioxide predisposes people to angiosarcomas and acute myeloid leukemia.
Radon, a radioactive gas that is released from soil, increases the risk of lung cancer, especially in people who smoke. Normally, radon disperses rapidly into the atmosphere and causes no harm. However, when a building is placed on soil with high radon content, radon can accumulate, sometimes producing sufficiently high levels in the air to cause harm.
Medications and chemicals
Combined estrogen and progestin oral contraceptives may slightly increase the risk of breast cancer, but this risk decreases over time. Estrogen therapy alone, without progesterone, increases the risk of uterine cancer. These increased risks are modest.
Diethylstilbestrol (DES) increases the risk of breast cancer in women who took the medication. It also increases the risk of vaginal cancer in women who were exposed before birth when their mother took DES.
Long-term use of anabolic steroids may increase the risk of liver cancer and accelerates prostate cancer.
Treatment of cancer with chemotherapy agents alone or with radiation therapy increases the risk of developing a second cancer, as do immunosuppressive agents given for organ transplantation. The cancers most commonly increased by immunosuppressants are skin cancers (including melanoma), kidney cancer, and neuroblastoma. Hydroxyurea increases the risk of skin cancer caused by sun exposure, in addition to increasing the risk of acute leukemia in patients with myeloproliferative disorders (eg, polycythemia vera, thrombocythemia) treated long-term with Treatment of cancer with chemotherapy agents alone or with radiation therapy increases the risk of developing a second cancer, as do immunosuppressive agents given for organ transplantation. The cancers most commonly increased by immunosuppressants are skin cancers (including melanoma), kidney cancer, and neuroblastoma. Hydroxyurea increases the risk of skin cancer caused by sun exposure, in addition to increasing the risk of acute leukemia in patients with myeloproliferative disorders (eg, polycythemia vera, thrombocythemia) treated long-term withhydroxyurea. Hydroxyurea not only causes 17p deletion, resulting in loss of a TP53 allele, it also impairs TP53 activation in patients with myeloproliferative disorders.
Chemical carcinogens can induce gene mutations and result in uncontrolled growth and tumor formation (see table ) (5). Other substances, called co-carcinogens, have little or no inherent carcinogenic potency but enhance the carcinogenic effect of another agent when exposed simultaneously.
Common Chemical Carcinogens
Carcinogens | Type of Cancer |
|---|---|
Environmental and industrial | |
Aromatic amines | |
Arsenic | |
Asbestos | Lung cancer |
Benzene | |
Chromates | Lung cancer |
Diesel exhaust | Lung cancer |
Formaldehyde | Leukemia Nasal cancer |
Hair dyes | Bladder cancer |
Ionizing radiation | Many types of cancer |
Manufactured mineral fibers | Lung cancer |
Nickel | Lung cancer Nasal sinus cancer |
Painting materials | Leukemia Lung cancer |
Pesticides, nonarsenic | Lung cancer |
Radon | Lung cancer |
Radiation | Most types of cancer |
Ultraviolet radiation | Skin cancer |
Vinyl chloride | Hepatic angiosarcoma |
Lifestyle | |
Alcohol | Mouth cancer Pharyngeal cancer |
Betel nuts | |
Tobacco | Bladder cancer Lung cancer |
Medications* | |
Alkylating agents (cyclophosphamide, platinum analogs) Alkylating agents (cyclophosphamide, platinum analogs) | Leukemia |
Diethylstilbestrol (DES) | Cervicovaginal cancer in women exposed in utero |
Immunosuppressants | Kaposi sarcoma Kidney cancer Lymphoma Skin cancer |
OxymetholoneOxymetholone | |
Topoisomerase inhibitors (anthracyclines, etoposide)Topoisomerase inhibitors (anthracyclines, etoposide) | Leukemia |
* Health care professionals exposed to antineoplastic agents are also at risk of adverse effects on reproduction. | |
Dietary substances
Certain substances consumed in the diet can increase the risk of cancer. For instance, a diet high in fat has been linked to an increased risk of colon, breast, and possibly prostate cancer. People who drink large amounts of alcohol are at higher risk of developing various cancers, including head and neck, liver, and esophageal cancer. A diet high in smoked and pickled foods or in meats cooked at a high temperature increases the risk of developing stomach cancer. People who have overweight or obesity have a higher risk of cancer of many cancer types, particularly cancers of the breast, endometrium, colon, kidney, and esophagus.
Physical factors
Chronic skin, lung, gastrointestinal, or thyroid inflammation may predispose to development of cancer. For example, patients with long-standing inflammatory bowel disease (ulcerative colitis) have an increased risk of colorectal carcinoma.
Sunlight and tanning light exposure increases the risk of skin cancers and melanoma.
Environmental factors references
1. White MK, Pagano JS, Khalili K. Viruses and human cancers: a long road of discovery of molecular paradigms. Clin Microbiol Rev 2014;27(3):463-481. doi:10.1128/CMR.00124-13
2. Lin WC, Tsai HF, Kuo SH, et al. Translocation of Helicobacter pylori CagA into Human B lymphocytes, the origin of mucosa-associated lymphoid tissue lymphoma. Cancer Res. 2010;70(14):5740-5748. doi:10.1158/0008-5472.CAN-09-46902.
3. Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med 1994;330(18):1267-1271. doi:10.1056/NEJM199405053301803
4. Botelho MC, Richter J. Editorial: Parasites and Cancer. Front Med (Lausanne). 2019;6:55. Published 2019 Mar 22. doi:10.3389/fmed.2019.00055
5. Baan R, Grosse Y, Straif K, et al. A review of human carcinogens--Part F: chemical agents and related occupations. Lancet Oncol 2009;10(12):1143-1144. doi:10.1016/s1470-2045(09)70358-4
Immune Disorders
Immune system dysfunction as a result of inherited genetic mutation, acquired disorders, aging, or immune-suppressing medications interferes with normal immune surveillance of early tumors and results in higher rates of cancer. Known cancer-associated immune disorders include
Ataxia-telangiectasia (acute lymphocytic leukemia [ALL], brain tumors, gastric cancer)
Wiskott-Aldrich syndrome (lymphoma, ALL)
X-linked agammaglobulinemia (lymphoma, ALL)
Immune deficiency resulting from immune-suppressing medications or HIV infection (lymphoma, kidney cancer, melanoma, cervical cancer, head and neck cancer, Kaposi sarcoma)
Autoimmune disorders such as systemic lupus erythematosus, rheumatoid arthritis, and Sjögren syndrome (B-cell lymphoma)
Drugs Mentioned In This Article





