



Platelets are circulating cell fragments that function in the clotting system. Thrombopoietin regulates platelet production by stimulating the proliferation and maturation of megakaryocytes in the bone marrow, which subsequently release platelets from their cytoplasm. Megakaryocytes are large, polyploid cells characterized by a multilobed nucleus with DNA content that can reach up to 64–128N (reflecting repeated endomitosis rather than multiple separate nuclei) and abundant cytoplasm. Through cytoplasmic fragmentation and proplatelet formation, each megakaryocyte can generate approximately 1,000 to 3,000 platelets. Thrombopoietin is produced in the liver at a constant rate; its circulating level is determined by how much is bound to circulating platelets and possibly to bone marrow megakaryocytes and the extent to which circulating platelets are cleared. Platelets circulate for 7 to 10 days. About one-third are always transiently sequestered in the spleen.
The platelet count is normally 140,000 to 440,000/mcL (140 to 440 × 109/L). However, the count can vary slightly according to menstrual cycle phase, decrease during near-term pregnancy (gestational thrombocytopenia), and increase in response to inflammatory cytokines (secondary, or reactive thrombocytosis). Platelets are eventually destroyed by apoptosis, a process independent of the spleen.
Platelet disorders include:
An abnormal increase in platelets (thrombocytosis)
A decrease in platelets (thrombocytopenia)
Platelet dysfunction
Thrombocytosis is a general term defined as an increase in the number of circulating platelets. That due to inflammatory cytokines is termed reactive or secondary thrombocytosis, and that due to clonal proliferation in the bone marrow is called primary thrombocytosis, thrombocythemia, or essential thrombocythemia. Although any of these conditions can predispose to thrombosis, even those in which platelets are markedly increased may also cause defective formation of hemostatic plugs and bleeding.
The risk of bleeding is inversely proportional to the platelet count and platelet function (see table ). When platelet function is reduced (eg, as a result of uremia or the use of a nonsteroidal anti-inflammatory drug [NSAID], aspirin, another medication, or a hereditary condition), the risk of bleeding increases.
Platelet Count and Bleeding Risk
Platelet Count | Risk of Bleeding |
|---|---|
≥ 50,000/mcL (≥ 50 × 109/L) | Minimal |
20,000–50,000/mcL (20–50 × 109/L) | Minor bleeding after trauma |
≤ 20,000/mcL (≤ 20 × 109/L) | Spontaneous bleeding |
≤ 5000/mcL (≤ 5 × 109/L) | Severe, possibly life-threatening spontaneous bleeding |
Etiology of Platelet Disorders
Thrombocythemia and thrombocytosis
Essential thrombocythemia is a myeloproliferative neoplasm involving overproduction of platelets because of a clonal abnormality of a hematopoietic stem cell. There is no correlation between the platelet count and risk of thrombosis, but some patients with extreme thrombocytosis (ie, > 1,000,000/mcL [> 1000 × 109/L]) develop bleeding due to loss of high molecular weight von Willebrand factor multimers (ie, acquired type 2B von Willebrand disease).
Reactive thrombocytosis is platelet overproduction in response to another disorder. There are many causes, including acute infection, chronic inflammatory disorders (eg, rheumatoid arthritis, inflammatory bowel disease, tuberculosis, sarcoidosis), iron deficiency, and certain cancers. Reactive thrombocytosis is not typically associated with an increased risk of thrombosis or bleeding.
Thrombocytopenia
Causes of thrombocytopenia can be classified by mechanism (see table ) and include:
Decreased platelet production
Increased splenic sequestration of platelets with normal platelet survival
Increased platelet destruction or consumption (both immunologic and nonimmunologic causes)
Dilution of platelets
Classification of Thrombocytopenia
Cause | Conditions |
|---|---|
Diminished or absent megakaryocytes in bone marrow | Amegakaryocytic thrombocytopenia Myelosuppressive medications (eg, hydroxyurea, interferon alfa-2b, chemotherapy agents) Paroxysmal nocturnal hemoglobinuria (some patients) |
Diminished platelet production despite the presence of megakaryocytes in bone marrow | Alcohol-induced thrombocytopenia Bortezomib use HIV-associated thrombocytopenia Myelodysplastic syndromes (some) |
Platelet sequestration in enlarged spleen | Cirrhosis with congestive splenomegaly |
Immunologic destruction | Systemic rheumatic diseases Hepatitis B-associated thrombocytopenia (uncommon) Hepatitis C-associated thrombocytopenia HIV-associated thrombocytopenia Influenza B Lymphoproliferative disorders (eg, chronic lymphocytic leukemia) Neonatal alloimmune thrombocytopenia Posttransfusion purpura |
Nonimmunologic destruction | Certain systemic infections (eg, hepatitis [any], infectious mononucleosis, cytomegalovirus infection, or dengue) Disseminated intravascular coagulation Thrombocytopenia in acute respiratory distress syndrome |
Dilution | Massive red blood cell replacement or exchange transfusion (most RBC transfusions use stored RBCs that do not contain many viable platelets) |
Unknown cause | Pregnancy (eg, gestational thrombocytopenia, HELLP syndrome [hemolysis, elevated liver enzymes, and low platelets])* † |
* Possible mechanisms may include increased destruction, decreased production, and placental sequestration. | |
† Additional information in Fogerty AE, Kuter DJ. How I Treat Thrombocytopenia in Pregnancy. Blood. 2024;143(9):747-756. doi:10.1182/blood.2023020726 | |
A large number of medications may cause thrombocytopenia, typically by triggering immunologic destruction.
Overall, the most common specific causes of thrombocytopenia include:
Pregnancy (gestational thrombocytopenia; HELLP syndrome [hemolysis, elevated liver enzymes, and low platelets]) (1)
Medications that cause immune-mediated platelet destruction (commonly, heparin, trimethoprim/sulfamethoxazole, rarely quinine [cocktail purpura] or abciximab), and rarely vaccinations (eg, influenza; shingles; measles, mumps, and rubella; COVID-19)
Medications and substances that cause dose-dependent bone marrow suppression (eg, chemotherapeutic agents, ethanol)
Systemic infection
Immune disorders (eg, immune thrombocytopenia [ITP], antiphospholipid syndrome, systemic lupus erythematosus)
Platelet dysfunction
Platelet dysfunction may stem from an intrinsic platelet defect or from an extrinsic factor that alters the function of normal platelets (1). Dysfunction may be hereditary or acquired. Hereditary disorders of platelet function consist of von Willebrand disease, the most common hereditary hemorrhagic disease, and inherited platelet function disorders, which are much less common. Acquired disorders of platelet dysfunction are commonly due to diseases (eg, renal failure) as well as to aspirin and other medications such as NSAIDs.
Etiology reference
1. Fogerty AE, Kuter DJ. How I Treat Thrombocytopenia in Pregnancy. Blood. 2024;143(9):747-756. doi:10.1182/blood.2023020726
Symptoms and Signs of Platelet Disorders
Platelet disorders result in a typical pattern of bleeding:
Multiple petechiae in the skin (typically most evident on the lower legs)
Scattered small ecchymoses at sites of minor trauma or venipuncture sites
Mucosal bleeding (oropharyngeal, nasal, gastrointestinal, genitourinary)
Excessive bleeding after surgery
Extensive menstrual bleeding

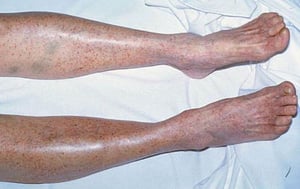
By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.

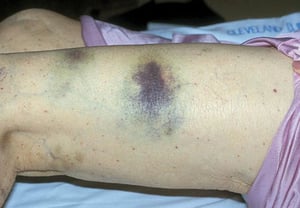
By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.

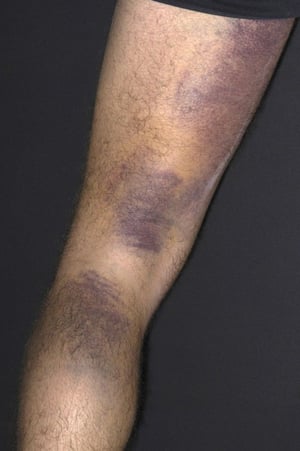
Ecchymoses are the large purple bruises seen on the leg of this patient.
Ecchymoses are the large purple bruises seen on the leg of this patient.
DR P. MARAZZI/SCIENCE PHOTO LIBRARY

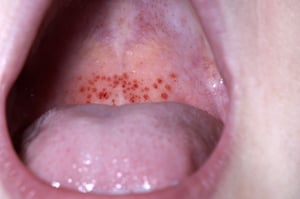
Petechiae are characterized by small red spots as seen here on the palate of this patient.
Petechiae are characterized by small red spots as seen here on the palate of this patient.
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
Ecchymoses are the large purple bruises seen on the leg of this patient.
Ecchymoses are the large purple bruises seen on the leg of this patient.
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
Petechiae are characterized by small red spots as seen here on the palate of this patient.
Petechiae are characterized by small red spots as seen here on the palate of this patient.
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
Heavy gastrointestinal bleeding and bleeding into the central nervous system are rare but may be life threatening. However, bleeding into tissues (eg, deep visceral hematomas or hemarthroses) rarely occurs with thrombocytopenia; instead, patients usually have immediate and superficial bleeding following an injury. Bleeding into the tissues (often delayed for up to a day after trauma) suggests a coagulation disorder (eg, hemophilia).
Diagnosis of Platelet Disorders
Clinical presentation of petechiae and mucosal bleeding
Complete blood count (CBC) with platelets, coagulation studies, peripheral blood smear
Sometimes bone marrow aspiration
Sometimes von Willebrand antigen, platelet-binding activity, and multimer studies
Sometimes platelet aggregation and platelet secretion assays
Platelet disorders are suspected in patients with petechiae and mucosal bleeding. A CBC with platelet count, coagulation studies, and a peripheral blood smear are obtained. Coagulation studies are normal unless there is a simultaneous coagulopathy. In patients with a normal CBC, platelet count, international normalized ratio (INR), and partial thromboplastin time (PTT), platelet or vessel wall dysfunction is suspected.
Pearls & Pitfalls
|
Thrombocytopenia
Peripheral smear examination is important in patients with thrombocytopenia because automated platelet counts sometimes show pseudothrombocytopenia due to platelet clumping caused by the ethylenediaminetetraacetic acid (EDTA) reagent present in most blood collection tubes. Also, schistocytes may be seen, which can indicate valvular hemolysis, thrombotic thrombocytopenic purpura (TTP), hemolytic-uremic syndrome (HUS), or disseminated intravascular coagulation (DIC—see table ).
Bone marrow aspiration is often indicated if the smear shows abnormalities other than thrombocytopenia, such as nucleated red blood cells (RBCs) or abnormal or immature white blood cells (WBCs). Bone marrow aspiration reveals the number and appearance of megakaryocytes and is the definitive test for many disorders that cause bone marrow failure. If the bone marrow is normal but the spleen is enlarged, increased splenic sequestration is the likely cause of thrombocytopenia. If the bone marrow is normal and the spleen is not enlarged, excess platelet destruction is the likely cause.
However, normal number and appearance of megakaryocytes do not always indicate normal platelet production. For example, in many patients with immune thrombocytopenia (ITP), platelet production may be decreased despite the normal appearance and increased number of megakaryocytes. In fact, bone marrow examination is rarely required in patients who present with typical features of immune thrombocytopenia.
The immature platelet fraction (IPF) in peripheral blood is sometimes a useful measure in patients with thrombocytopenia because it is elevated when the bone marrow is producing platelets and not increased when bone marrow platelet production is reduced, similar to the reticulocyte count in anemia. Equally important is assessment of platelet size by microscopic examination or measurement of the mean platelet volume (MPV); in thrombocytopenic disorders of increased platelet turnover, larger platelets are produced.
Measurement of antiplatelet antibodies and thrombopoietin (TPO) level may be clinically useful in some patients to distinguish ITP from other causes of thrombocytopenia (1). In patients with ITP, the antiplatelet antibody is often positive and the TPO normal, whereas in disorders of reduced platelet production, the antiplatelet antibody test is usually negative with elevated TPO level. HIV testing is done in patients with or at risk of HIV infection, hepatitis B infection or hepatitis C infection, or HIV and hepatitis coinfection.
Peripheral Blood Findings in Thrombocytopenic Disorders
Finding | Disorders |
|---|---|
Normal red blood cells (RBCs) and white blood cells (WBCs) | Drug-induced thrombocytopenia Hepatitis C–related thrombocytopenia HIV-related thrombocytopenia Posttransfusion purpura |
RBC fragmentation (schistocytes) | Disseminated intravascular coagulation (DIC) HELLP syndrome (hemolytic anemia, elevated liver enzymes, low platelets) Metastatic cancer to the bone marrow (ie, myelophthisis) Preeclampsia with DIC Thrombotic thrombocytopenic purpura Valvular hemolysis |
WBC abnormalities | Hypersegmented polymorphonuclear leukocytes in megaloblastic anemias Atypical lymphocytes in large granular lymphocyte leukemia or increased mature lymphocytes in chronic lymphocytic leukemia Markedly diminished granulocytes in aplastic anemia Blasts and other immature cells in acute leukemia (myeloid/lymphoblastic/mixed phenotype) |
Frequent giant platelets (approaching the size of RBCs) | Bernard-Soulier syndrome Disorders related to the myosin heavy chain 9, non-muscle gene (MYH9) Other congenital thrombocytopenias Myelodysplastic syndrome (MDS) |
RBC abnormalities, nucleated RBCs, and immature granulocytes | Myelofibrosis Metastatic cancer to the bone marrow (ie, myelophthisis) |
Platelet clumping | Pseudothrombocytopenia |

Schistocytes (see arrows) are damaged red blood cells, which may occur in microangiopathic hemolytic anemia (including disseminated intravascular coagulation, thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome, and valvular hemolysis).
By permission of the publisher. From Tefferi A, Li C. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
Suspected platelet dysfunction
In patients with suspected platelet dysfunction, a medication should be considered as a possible cause when symptoms begin after initiation of an agent known to affect platelets. Common culprits include antiplatelet agents (eg, aspirin, clopidogrel, prasugrel, ticagrelor, cangrelor, abciximab, eptifibatide, tirofiban), as well as other medications that can impair platelet function such as nonsteroidal anti-inflammatory drugs (NSAIDs), selective serotonin reuptake inhibitors (SSRIs), valproic acid, and certain antibiotics (eg, beta-lactams). Platelet dysfunction caused by medications may be severe, but specialized tests are rarely needed.
A hereditary cause (2) is suspected if there is a lifelong history of easy bruising; bleeding after tooth extractions, surgery, childbirth, or circumcision; or heavy menstruation. In the case of a suspected hereditary cause, von Willebrand factor (VWF) antigen and VWF activity studies are routinely done.
In patients with suspected hereditary dysfunction, platelet aggregation and secretion tests may identify a defect in how the platelet responds to various platelet agonists (adenosine diphosphate [ADP], collagen, ristocetin, thrombin) and thereby demonstrate the type of platelet defect.
Platelet dysfunction caused by most systemic disorders is typically mild and of minor clinical importance; in these patients, the causative systemic disorder is the clinical concern, and hematologic tests are unnecessary. However, patients with renal failure may develop significant bleeding.
Diagnosis references
1. Hayward CPM, Tasneem S. When it's not Glanzmann thrombasthenia or Bernard-Soulier syndrome: diagnosing other qualitative platelet disorders. Hematology Am Soc Hematol Educ Program. 2025;2025(1):137-146. doi:10.1182/hematology.2025000699
2. Al-Samkari H, Rosovsky RP, Karp Leaf RS. A modern reassessment of glycoprotein-specific direct platelet autoantibody testing in immune thrombocytopenia. Blood Adv. 2020;4(1):9-18. doi: 10.1182/bloodadvances.2019000868
Treatment of Platelet Disorders
Stopping medications that impair platelet function
Thrombopoietin receptor agonists (TPO-RA)
Sometimes platelet transfusions
Sometimes antifibrinolytic agents
Rarely splenectomy
In patients with thrombocytopenia or platelet dysfunction, medications that further impair platelet function, particularly aspirin and NSAIDs, should not be given. In patients who are already taking such medications, alternatives, such as acetaminophen, should be considered.
Patients may require platelet transfusion, but transfusions are given only in limited situations. For example, platelet transfusion is the mainstay of therapy for patients with platelet dysfunction and active bleeding or for those in need of an invasive procedure. Prophylactic transfusions are used sparingly because they may lose their effectiveness with repeated use due to the development of platelet alloantibodies (1). Recent evidence and guidelines provide different recommendations regarding prophylactic platelet transfusions in nonbleeding patients for some conditions (eg, induction and consolidation phases of treatment for acute leukemia) at different levels of decreased platelet count (1).
If decreased production is the cause of thrombocytopenia, transfusions, TPO-RA (eg, romiplostim, eltrombopag, avatrombopag, hetrombopag) (2), or antifibrinolytic agents (eg, aminocaproic acid, tranexamic acid) are generally reserved for patients with any of the following:
Active bleeding
Severe thrombocytopenia (eg, platelet count < 10,000/mcL [< 10 × 109/L)
A need for an invasive procedure
Although antifibrinolytics such as aminocaproic acid and tranexamic acid are sometimes used as adjuncts in patients with thrombocytopenia who are bleeding, current evidence does not support their routine prophylactic use in hypoproliferative thrombocytopenia (3).
If platelet destruction is the cause of thrombocytopenia, transfusions are reserved for life-threatening, central nervous system, or ocular bleeding (4). Other options include splenectomy, immunosuppressive agents (including glucocorticoids), TPO-RA, rilzabrutinib (a Bruton tyrosine kinase [BTK] inhibitor) (5, 6), and fostamatinib (a spleen tyrosine kinase inhibitor) (7).
Treatment references
1. Metcalf RA, Nahirniak S, Guyatt G, et al. Platelet Transfusion: 2025 AABB and ICTMG International Clinical Practice Guidelines. JAMA. 2025;334(7):606-617. doi:10.1001/jama.2025.7529
2. Gauer RL, Whitaker DJ. Thrombocytopenia: Evaluation and Management. Am Fam Physician. 2022;106(3):288-298.
3. Champaneria R, Estcourt LJ, Geneen L, et al. Antifibrinolytics (lysine analogues) for the prevention of bleeding in people with haematological disorders. Cochrane Database Syst Rev. 2026;2:CD009733. doi:10.1002/14651858.CD009733.pub4
4. Chowdhury SR, Sirotich E, Guyatt GH, et al. Guideline on the emergency management of critical bleeding in patients with immune thrombocytopenia. Blood Adv. Published online February 4, 2026. doi:10.1182/bloodadvances.2025018818
5. Kuter DJ, Efraim M, Mayer J, et al. Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia. N Engl J Med. 2022;386(15):1421-1431. doi:10.1056/NEJMoa2110297
6. Kuter DJ, Ghanima W, Cooper N, et al. Safety and efficacy of rilzabrutinib vs placebo in adults with immune thrombocytopenia: the phase 3 LUNA3 study. Blood. 2025;145(24):2914-2926. doi:10.1182/blood.2024027336
7. González-López TJ, Bermejo-Vega N, Cardesa-Cabrera R, et al. Fostamatinib effectiveness and safety for immune thrombocytopenia in clinical practice. Blood. 2024;144(6):646-656. doi:10.1182/blood.2024024250
Drug Information for the Topic





