



Cerebral hemispheres with congenital malformations may be large, small, or asymmetric; the gyri may be absent, unusually large, or multiple and small.
In addition to the grossly visible malformations, microscopic sections of normal-appearing brain may show disorganization of the normal laminar neuronal arrangement. Localized deposits of gray matter may be present in regions normally occupied only by white matter (heterotopic gray matter).
Malformations of the cerebral hemispheres may be due to genetic or acquired causes. Acquired causes include infections (eg, cytomegalovirus, Zika virus infection) and vascular or metabolic causes that interrupt the blood or nutrient supply to the developing brain (eg, in utero stroke, maternal diabetes or illicit drug use).
Microcephaly or macrocephaly, moderate to severe motor and intellectual disability, and epilepsy often occur with these defects, with highly variable manifestations.
Prenatally, diagnosis is by ultrasound, MRI, or both. However, a significant proportion of subtle malformations can be missed by prenatal imaging. Postnatally, diagnosis is largely based on the physical and neurologic findings described above.
Treatment is supportive, including antiseizure medications, specialized education, and, if needed, therapy.
Holoprosencephaly
Holoprosencephaly spectrum occurs when the embryonic prosencephalon (which becomes the forebrain) does not undergo complete segmentation and cleavage.
Holoprosencephalies may be caused by mutations in a number of genes, of which > 14 are known; among these are the sonic hedgehog signaling pathway genes and their modifiers (1). Trisomy 13 and trisomy 18, as well as other chromosomal deletions and duplications, have been associated with holoprosencephaly.
The 3 main types of holoprosencephaly, in declining order of severity, are:
Alobar
Semilobar
Lobar
Alobar holoprosencephaly is the most severe and is usually fatal. It is characterized by complete failure of cleavage and a single ventricular cavity without any septation.
Semilobar holoprosencephaly is characterized by partial cleavage into hemispheres posteriorly but with a communicating unified ventricular cavity anteriorly.
Lobar holoprosencephaly is characterized by absence of the septum pellucidum (the membrane that separates the front of the 2 lateral ventricles), agenesis of the corpus callosum, fusion of the anterior horns of the lateral ventricles, and possibly fusion of the cingulate gyri.
A fourth, rare type, called middle interhemispheric variant, is characterized by fusion of the posterior frontal and parietal lobes as well as possibly the thalamus but with normal hemispheric differentiation elsewhere.
Rhombencephalosynapsis is a malformation similar to holoprosencephaly but mainly involves the hindbrain (2). In rhombencephalosynapsis, there is fusion of the cerebellar hemispheres with partial or complete absence of the vermis (the midline portion of the cerebellum). This malformation can result in aqueductal stenosis and hydrocephalus. Other possible associated abnormalities include forebrain holoprosencephaly, absence of the olfactory bulbs, dysgenesis of the corpus callosum or septum pellucidum, and VACTERL (vertebral anomalies, anal atresia, cardiac anomalies, tracheoesophageal fistula, renal anomalies, and limb anomalies).
Craniofacial anomalies are present in the holoprosencephalies, and their degree of severity usually mirrors the underlying brain abnormality. Included in the spectrum are anophthalmia or cyclopia, malformed or absent nares or nasal cavity, hypotelorism, cleft lip and cleft palate, and a central incisor.
Severely affected fetuses may die in utero. After birth, manifestations include seizures, intellectual disability, low muscle tone, and motor delays that affect all modalities of functioning.
Treatment of holoprosencephaly is supportive.
Holoprosencephaly references
1. Hong M, Srivastava K, Kim S, et al: BOC is a modifier gene in holoprosencephaly. Hum Mutat 38(11):1464–1470, 2017. doi: 10.1002/humu.23286
2. Ishak G, Dempsey J, Shaw D, et al: Rhombencephalosynapsis: A hindbrain malformation associated with incomplete separation of midbrain and forebrain, hydrocephalus and a broad spectrum of severity. Brain 135(5):1370–1386, 2012. doi: 10.1093/brain/aws065
Lissencephaly
Lissencephaly consists of an abnormally thick cortex, diminished or absent gyral pattern on the surface of the brain, reduced or abnormal lamination of the cerebral cortex, and often diffuse neuronal heterotopias.
This malformation is caused by abnormal neuronal migration, the process by which immature neurons attach to radial glia and move from their points of origin near the ventricle to the cerebral surface. Several single-gene defects may cause this anomaly (eg, LIS1). One X-linked gene, DCX, causes familial X-linked lissencephaly in males and another, generally milder, migrational abnormality in females called subcortical band heterotopia. In subcortical band heterotopia, a broad swath of ectopic gray matter in the subcortical white matter resembles a "double cortex" on MRI.
Affected infants almost always have significant intellectual disability and seizures (often infantile spasms).
Treatment of lissencephaly is supportive.
Survival depends on seizure severity and the presence of other complications including swallowing dysfunction, apnea, and difficulty clearing oropharyngeal secretions.
Polymicrogyria
Polymicrogyria, in which the gyri are small and overabundant, also involves abnormal neuronal migration. Other common findings include simplified or absent cortical lamination in affected regions, heterotopic gray matter, a hypoplastic or absent corpus callosum and septum pellucidum, and malformations of the brain stem and/or cerebellum.
The structural abnormalities may be diffuse or focal. The most common area of focal involvement is the perisylvian fissure (bilaterally or unilaterally). The term perisylvian syndrome is sometimes used when children present with features of epilepsy, facial and oral motor weakness, prominent speech and language delays, and usually bilateral polymicrogyria in the sylvian fissure region.
Polymicrogyria is highly associated with schizencephaly, in which there are abnormal slits, or clefts, in the cerebral hemispheres.
Numerous causes of polymicrogyria have been identified, including a number of single-gene mutations (eg, of SRPX2), and primary maternal infection with cytomegalovirus (ie, in which the mother has no prior immunity—see Congenital and Perinatal Cytomegalovirus Infection).
The most common clinical manifestations are seizures, intellectual disability, and spastic hemiplegia or diplegia (see also Schizencephaly).

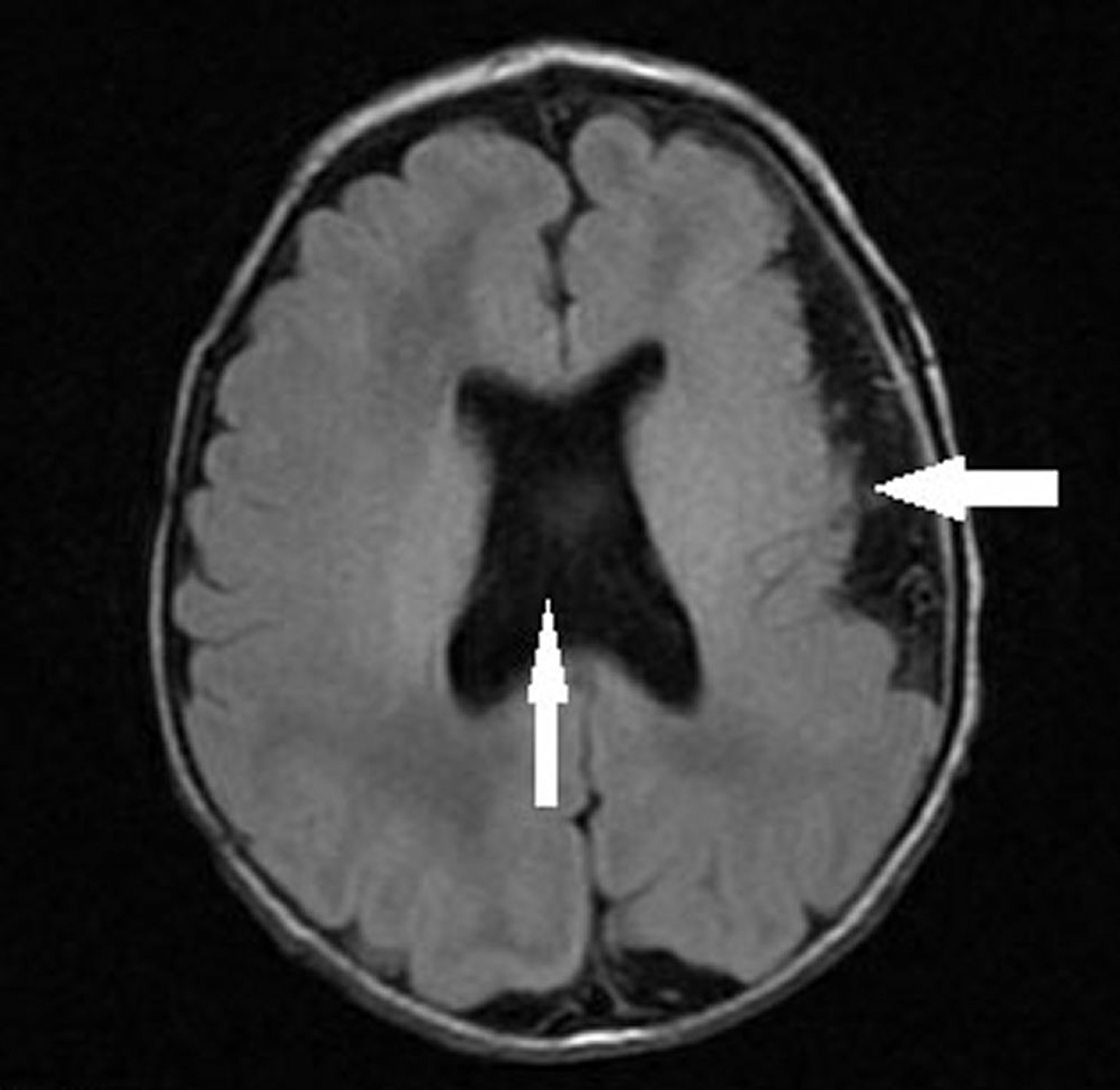
This image shows an infant with left hemispheric polymicrogyria (leftward arrow) and absent septum pellucidum with abnormal ventricular configuration (vertical arrow) consistent with septo-optic dysplasia (optic nerve hypoplasia, absent or abnormal septum pellucidum, and pituitary hypoplasia).
Image courtesy of Stephen J. Falchek, MD.
Treatment of polymicrogyria is supportive.





