







Les lymphomes non hodgkiniens regroupent un ensemble hétérogène de pathologies impliquant une prolifération monoclonale maligne de cellules lymphoïdes du système lymphoréticulaire, incluant les aires ganglionnaires, la moelle osseuse, la rate, le foie et le tractus respiratoire. La symptomatologie initiale comprend habituellement la présence d'une adénopathie périphérique. Cependant, certains patients n'ont pas d'adénopathie à la présentation, mais ont des lymphocytes anormaux dans le sang périphérique. La maladie est susceptible d'être disséminée au moment de la présentation et le diagnostic repose généralement sur la biopsie des ganglions lymphatiques et/ou de la moelle osseuse. Les stratégies de prise en charge peuvent comprendre la surveillance et l'attente, la chimiothérapie, les médicaments ciblés (p. ex., les inhibiteurs de kinase) et les immunothérapies (p. ex., anticorps monoclonaux, CAR-T [chimeric antigen receptor T cells]); parfois, une radiothérapie est ajoutée. À quelques exceptions près, la transplantation de cellules-souches est habituellement réservée aux patients présentant des lymphomes agressifs après une rémission incomplète ou une rechute.
(Voir aussi Revue générale des lymphomes.)
Les lymphomes non hodgkiniens sont plus fréquents que les lymphomes d'Hodgkin. C'est le 6e cancer par ordre de fréquence aux États-Unis et représente 4% de tous les nouveaux cancers aux États-Unis chaque année et 3% de tous les décès par cancer. Plus de 80 000 nouveaux cas sont diagnostiqués chaque année dans tous les groupes d'âge avec a environ 20 000 décès (1). Les lymphomes non hodgkiniens ne représentent pas une entité unique, mais plutôt un ensemble de pathologies malignes des lymphocytes avec un certain nombre de sous-groupes subdivisés en types agressifs et indolents. La prévalence augmente avec l'âge.
Référence générale
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73(1):17-48. doi:10.3322/caac.21763
Étiologie des lymphomes non hodgkiniens
La cause des lymphomes non hodgkiniens est inconnue, bien que, comme pour les leucémies, il existe des arguments en faveur d'une origine virale (p. ex., Human T-cell Leukemia-lymphoma virus, virus Epstein-Barr, virus de l'hépatite B, virus de l'hépatite C, VIH, human herpesvirus 8) dans certains cas. L'infection à Helicobacter pylori augmente également le risque de lymphome.
Les patients présentant un risque accru de lymphome non hodgkinien comprennent ceux qui ont un
Déficit immunitaire secondaire (p. ex., Lorsqu'il est induit par des immunosuppresseurs, tels que ceux utilisés dans les maladies rhumatismales systémiques et après une transplantation d'organe solide)
Pathologies auto-immunes (p. ex., polyarthrite rhumatoïde, syndrome de Sjögren)
Inflammation chronique et hyperplasie réactive des ganglions lymphatiques
Éventuellement exposition à certains produits chimiques (p. ex., certains herbicides et insecticides)
Le lymphome non hodgkinien est l'un des cancers les plus fréquents chez les patients infectés par le VIH et certains patients infectés par le VIH se présentent initialement avec un lymphome. Les patients qui ont un lymphome non hodgkinien doivent généralement être dépistés pour le VIH et les virus de l'hépatite.
Les facteurs génétiques semblent avoir un rôle. Certains polymorphismes mononucléotidiques augmentent le risque de lymphome. Les patients ayant un parent au premier degré atteint d'un lymphome hodgkinien ou non hodgkinien présentent un risque accru de lymphome non hodgkinien.
Physiopathologie des lymphomes non hodgkiniens
La plupart des lymphomes non hodgkiniens proviennent des lymphocytes B; les autres lymphomes non hodgkiniens proviennent des lymphocytes T ou des cellules NK (Natural Killer). Le stade de la différenciation lymphocytaire à laquelle se produit l'événement oncogène détermine la présentation et l'évolution de la maladie.
La plupart des lymphomes sont nodaux avec une atteinte variable de la moelle osseuse et du sang périphérique, bien que certains lymphomes apparaissent ou impliquent des sites extranodaux (p. ex., peau, tractus gastro-intestinal, poumon, système nerveux central). Un tableau clinique proche d'une leucémie, avec hyperlymphocytose périphérique et atteinte médullaire, peut être retrouvé jusque dans 50% des formes de l'enfant et environ 20% de certaines formes de l'adulte de lymphome non hodgkinien.
Une hypogammaglobulinémie provoquée par une diminution progressive de la production d'immunoglobulines est présente chez 15% des patients au moment du diagnostic. L'hypogammaglobulinémie augmente le risque d'infection bactérienne grave et les patients peuvent avoir besoin d'immunoglobulines IV pour compenser le déficit en immunoglobulines.
Pièges à éviter
|
Classification des lymphomes non hodgkiniens
La classification anatomopathologique des lymphomes non hodgkiniens continue à évoluer, du fait de l'évolution des connaissances nouvelles sur les cellules à l'origine de la maladie et les bases biologiques de ce groupe hétérogène. Depuis 2022, 2 systèmes de classification existent: la classification de l'OMS de 2022 (1) et l'International Consensus Classification de 2022 (2). Les deux systèmes intègrent des éléments de morphologie, d'immunophénotype, d'information génétique et de profils cliniques pour subdiviser et classer les maladies en entités distinctes présentant un intérêt clinique.
On distingue généralement les lymphomes non hodgkiniens indolents des lymphomes agressifs:
Lymphomes indolents: ils évoluent lentement, répondent lentement au traitement, mais ne sont pas généralement curables par les approches thérapeutiques standards
Agressifs: rapidement évolutifs, mais très sensibles à la chimiothérapie et souvent curables
Chez l'enfant, les lymphomes non hodgkiniens sont presque toujours agressifs. Les lymphomes folliculaires et les autres lymphomes de bas grade sont inhabituels. Le traitement des lymphomes agressifs (lymphome de Burkitt, lymphome diffus à grands lymphocytes B et lymphome lymphoblastique) pose des problèmes spécifiques, notamment du fait de l'atteinte gastro-intestinale (en particulier de l'iléon terminal); de la diffusion méningée (nécessitant un traitement ou une prophylaxie du liquide céphalorachidien); et des autres sites sanctuaires de la maladie (p. ex., testicules, cerveau). En outre, ces lymphomes étant potentiellement curables, l'impact des effets indésirables et le pronostic doivent être pris en compte, en particulier les risques de cancer secondaire tardif, les séquelles cardiorespiratoires, la préservation de la fertilité et les conséquences sur le développement.
Références sur la classification
1. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms [published correction appears in Leukemia 2023 Sep;37(9):1944-1951]. Leukemia 2022;36(7):1720-1748. doi:10.1038/s41375-022-01620-2
2. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee [published correction appears in Blood 2023 Jan 26;141(4):437]. Blood 2022;140(11):1229-1253. doi:10.1182/blood.2022015851
Symptomatologie des lymphomes non hodgkiniens
La plupart des patients ont au début un des signes suivants
Lymphadénopathie périphérique asymptomatique
Les ganglions augmentés de volumes peuvent être élastiques, isolés initialement et secondairement se fondent par la suite en masses. Les ganglions affectés ne sont généralement pas douloureux, contrairement aux ganglions des infections virales. L'atteinte ganglionnaire est localisée chez certains patients, mais, dans la plupart des cas, elle est multifocale. L'examen clinique initial doit rechercher soigneusement des ganglions cervicaux, axillaires, inguinaux et fémoraux.
Chez certains patients, des ganglions médiastinaux et rétropéritonéaux augmentés de pression exercent une pression sur les structures voisines, ce qui entraîne des symptômes. Les plus importants d'entre eux sont
Compression de la veine cave supérieure: essoufflement et œdème facial (Syndrome cave supérieur)
Compression de l'arbre biliaire externe: ictère
Compression des uretères: hydronéphrose
Occlusion intestinale: vomissements et constipation
Perturbation du drainage lymphatique: liquide pleural ou péritonéal chyleux ou lymphœdème d'un membre inférieur
La peau est atteinte dans certains lymphomes non hodgkiniens. Les lymphomes non hodgkiniens à lymphocytes B peuvent affecter le cuir chevelu (lymphomes non hodgkiniens folliculaires) ou les jambes (grosses cellules), causant généralement des nodules érythémateux légèrement surélevés. Dans les lymphomes non hodgkiniens à lymphocytes T cutanés, les lésions cutanées peuvent consister en des tumeurs, des plaques ou des papules ou un érythème diffus non palpable ou des papules discrètes. Chez les patients qui ont une peau foncée, l'érythème peut être peu visible.

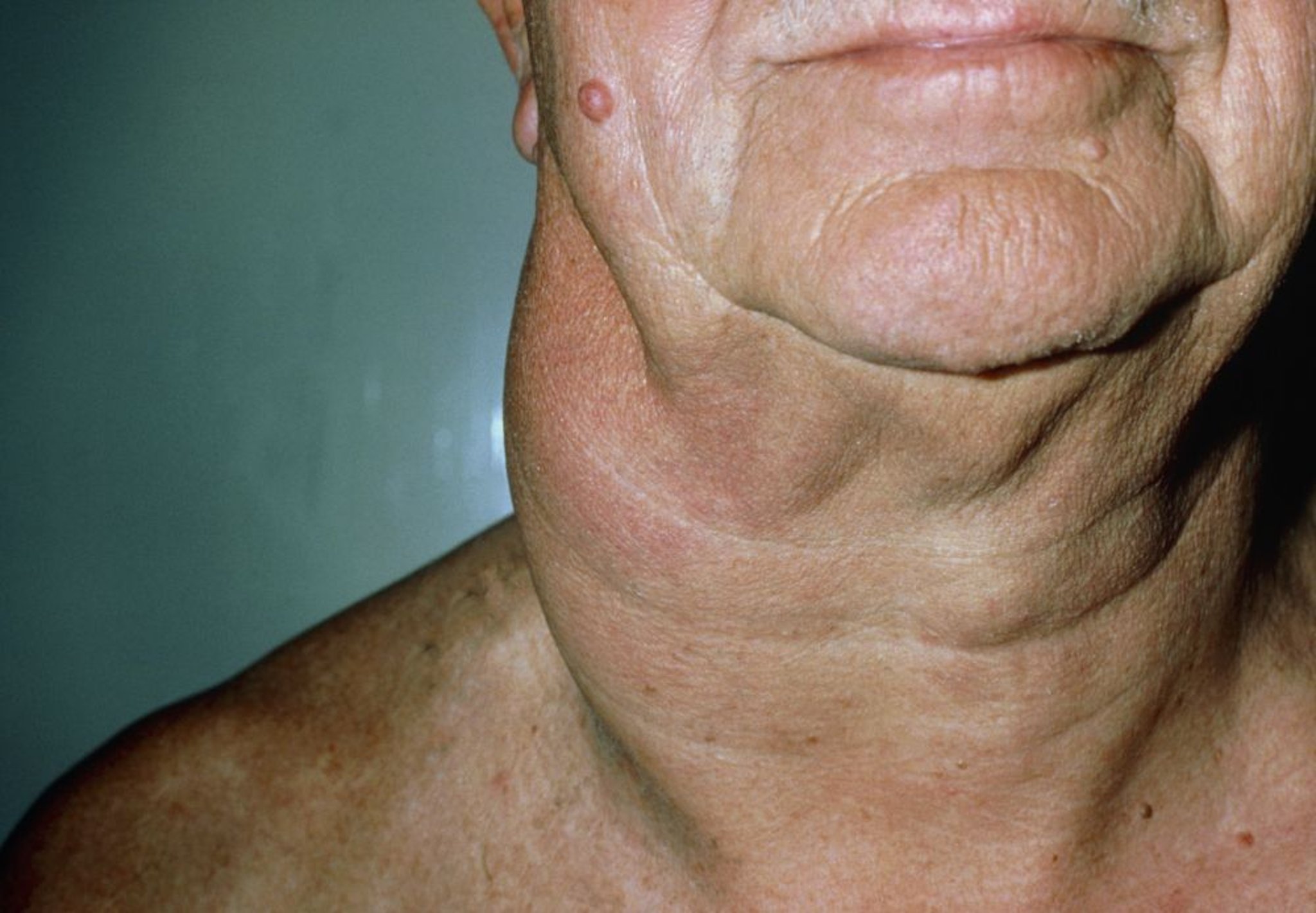
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
Les symptômes systémiques (p. ex., fatigue, fièvres, sueurs nocturnes, perte de poids) peuvent être les premières manifestations chez certains patients, le plus souvent dans les lymphomes agressifs. Ces patients peuvent ne pas avoir remarqué de lymphadénopathie ou ne pas avoir de maladie externe palpable; ces patients nécessitent une imagerie TDM ou PET (positron emission tomography) scan pour mettre en évidence la ou les lésion(s).

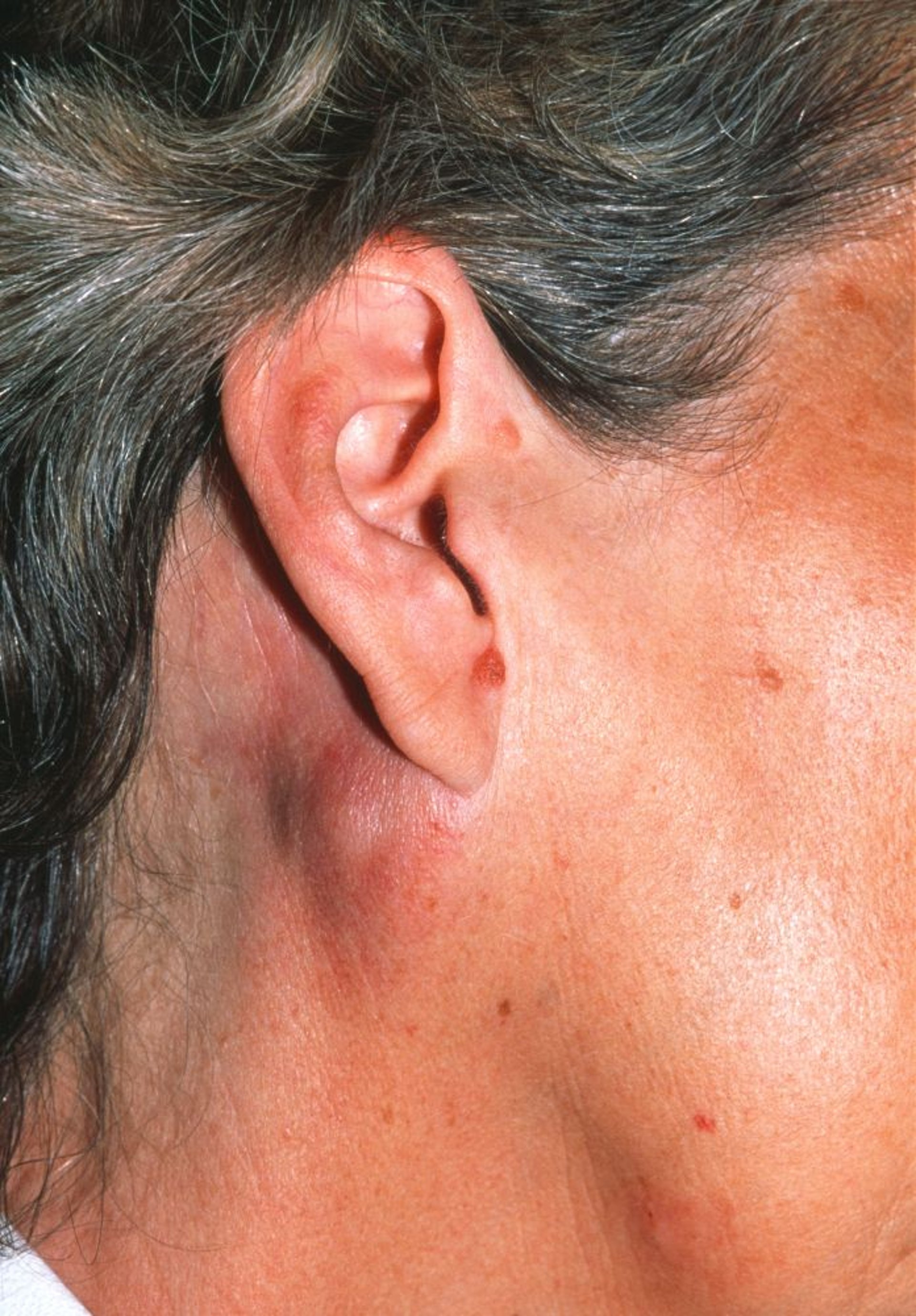
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
Une anémie est présente initialement chez certains patients et se développe finalement dans de nombreux cas. Elle peut être provoquée par
Un saignement dû à un lymphome gastro-intestinal, avec ou sans hypoplaquettose
Une hémolyse due à un hypersplénisme ou à une anémie hémolytique à Coombs positif
Une infiltration de la moelle osseuse due à un lymphome
La suppression de la moelle osseuse par la chimiothérapie ou la radiothérapie
La suppression de la fonction de la moelle osseuse liée à une inflammation chronique
Manifestations de certains lymphomes particuliers
Les lymphomes/leucémies T de l'adulte (associés à HTLV-1 [Human T-lymphotrophic virus-1) ont une évolution clinique fulminante, avec infiltration cutanée, lymphadénopathies, hépatosplénomégalie et leucémie. Les cellules leucémiques des lymphocytes T malins qui ont très fréquemment des noyaux multilobés. Une hypercalcémie est fréquente, plus liée à des facteurs humoraux qu'à un envahissement osseux direct.
Les lymphomes anaplasiques à grandes cellules peuvent causer des lésions cutanées, des adénopathies et des lésions viscérales rapidement progressives. Cette maladie peut être confondue avec un lymphome d'Hodgkin ou un carcinome métastatique non différencié.
Diagnostic des lymphomes non hodgkiniens
Biopsie de ganglion lymphatique
Souvent aspiration et biopsie unilatérales de moelle osseuse
FDG-PET/TDM thoracique et abdomino-pelvienne pour la classification par stades
IRM du cerveau et/ou de la moelle épinière si des symptômes neurologiques sont présents
Comme dans le lymphome d'Hodgkin, le diagnostic de lymphome non hodgkinien est habituellement évoqué devant
Lymphadénopathie indolore
Adénopathie détectée sur une radiographie pulmonaire ou une TDM effectuée pour d'autres raisons
La présence d'une adénopathie indolore peut également faire évoquer une mononucléose infectieuse, une toxoplasmose, une infection à cytomégalovirus, une primo-infection à VIH ou une leucémie aiguë.
Les anomalies de la rx thorax peuvent mimer un carcinome pulmonaire, une sarcoïdose ou une tuberculose.
Moins fréquemment, les patients sont diagnostiqués devant une hyperlymphocytose retrouvée sur une NFS réalisée pour des symptômes non spécifiques. Dans ces circonstances, le diagnostic différentiel inclut les leucémies, une infection due au virus Epstein-Barr ou un syndrome de Duncan (syndrome lymphoprolifératif lié à l'X).
Les tests nécessaires pour établir le diagnostic sont suivis de tests pour compléter la classification par stade et évaluer l'étiologie et le pronostic (1).
Tests diagnostiques
Les ganglions lymphatiques augmentés de volume sont biopsiés. Si un ganglion est palpable, aucune imagerie n'est nécessaire initialement, bien qu'une TDM ou une échographie puissent être nécessaires pour planifier correctement les tests ultérieurs.
Si la lésion est facilement palpable, une biopsie ouverte est préférable. Si la lésion est située dans le poumon ou l'abdomen, une biopsie au trocart (de calibre 18 à 20) réalisée sous guidage TDM ou échographique peut souvent obtenir un prélèvement permettant le diagnostic. Une biopsie à l'aiguille fine (percutanée ou bronchoscopique) ne produira souvent pas de tissu suffisant, en particulier pour le diagnostic initial; la biopsie par forage est préférée si elle est jugée sans danger.
Les biopsies doivent être examinées par un pathologiste spécialisé dans le diagnostic du lymphome afin que le lymphome puisse être correctement classé. Si cette révision n'est pas disponible localement, les lames doivent être envoyées à un laboratoire de référence ayant une expertise en hématopathologie. La classification correcte du lymphome non hodgkinien est essentielle à la planification du traitement. Les lymphomes non hodgkiniens sont potentiellement curables, mais sans diagnostic précis, un traitement optimal peut ne pas être choisi.
Les critères histologiques sur la biopsie sont la destruction de l'architecture normale des ganglions lymphatiques et un envahissement de la capsule et de la graisse adjacente par les cellules malignes.
Les études d'immunophénotypage (par immunohistochimie ou cytométrie de flux) visant à déterminer l'origine cellulaire caractérisent le sous-type spécifique et permettent d'évaluer le pronostic et de définir la prise en charge thérapeutique; ces études peuvent aussi être faites sur des cellules sanguines si elles sont présentes, mais généralement ces colorations sont appliquées sur du tissu fixé au formol et inclus dans de la paraffine.
La mise en évidence de l'antigène leucocytaire commun CD45 par la méthode d'immunoperoxydase élimine le diagnostic de carcinome métastatique, qui fait souvent partie des diagnostics différentiels "des cancers indifférenciés". La recherche de l'antigène leucocytaire commun, l'identification de la plupart des marqueurs de surface et des réarrangements géniques (pour documenter la clonalité des lymphomes B ou T) peuvent être réalisés sur tissus fixés. La cytogénétique et la cytométrie en flux nécessitent du tissu frais.
Le séquençage de nouvelle génération peut avoir une signification diagnostique ou pronostique en cas de lymphome non hodgkinien et peut être effectué sur des tissus frais ou fixés (dépendant du test).
Examens de classification par stades
Une fois le diagnostic de lymphome posé, des examens de classification par stades sont effectués.
Une PET-FDG (fluorodésoxyglucose)-/TDM combinée du thorax, de l'abdomen et du bassin est recommandée. La PET/TDM permet de localiser avec précision les lésions, leur taille (par la TDM) et le métabolisme de la tumeur (par la FDG-PET). Si la FDG-PET/TDM n'est pas disponible, une TDM du thorax, de l'abdomen et du bassin est effectué.
Un myélogramme et une biopsie de moelle osseuse unilatérales sont souvent effectués chez presque tous les patients qui ont un lymphome non hodgkinien. Bien que l'évaluation de la moelle puisse avoir un intérêt diagnostique, son utilité pour définir le stade et le pronostic de la plupart des lymphomes est moins évidente. L'évaluation de la moelle osseuse peut être d'une utilité limitée dans les situations où l'atteinte médullaire est peu probable (p. ex., lymphome diffus à grandes cellules B précoce) ou dans les situations où les résultats n'influenceraient probablement pas la prise en charge (p. ex., maladie à un stade avancé).
Exploration destinées aux complications et pronostic
Les tests sanguins comprennent généralement une NFS, un comptage des GB, les tests de la fonction rénale et hépatique (dont la créatinine sérique, la bilirubine, le calcium, l'AST (aspartate aminotransférase), l'albumine, la phosphatase alcaline et la lactate déshydrogénase), l'acide urique, la bêta-2 microglobuline et la vitamine D. Une électrophorèse des protéines sériques avec taux d'IgG, d'IgA et d'IgM est également effectuée.
D'autres examens biologiques sont effectués en fonction des signes (p. ex., IRM du cerveau et/ou de la moelle épinière en cas de symptômes neurologiques). Si les taux d'acide urique sont élevés, le taux de G6PD sérique doit être vérifié car le déficit en G6PD (glucose-6-phosphate déshydrogénase) exclut un traitement par la rasburicase qui est souvent administré pour prévenir un syndrome de lyse tumorale mais peut provoquer une anémie hémolytique en cas de déficit en G6PD.
Test de l'étiologie
Les patients qui ont un lymphome non hodgkinien doivent être dépistés à la recherche du HIV et de l'hépatite B et C. Les patients chez qui on a diagnostiqué une leucémie/lymphome à cellules T adultes (ATLL) sont également testés à la recherche du virus lymphotrope T de type 1 humain (human T-cell lymphotropic virus type 1, HTLV-1).
Classification par stades
Une fois le diagnostic établi, le stade est déterminé afin de guider le traitement. Le système de classification par stades de Lugano couramment utilisé (voir tableau Classification par stades de Lugano du lymphome hodgkinien et des lymphomes non hodgkiniens) intègre
Symptômes
Résultats de l'examen clinique
L'imagerie, dont la rx thorax, la TDM du thorax, et abdomino-pelvienne et la PET-FDG
Biopsie de moelle osseuse (dans certains cas)
Bien que des formes de lymphome non hodgkinien (stade I) existent, la maladie est habituellement disséminée lors de sa découverte.
Référence pour le diagnostic
1. Cheson BD, Fisher RI, Barrington SF, et al: Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J Clin Oncol 32(27):3059–3068, 2014.
Traitement des lymphomes non hodgkiniens
Surveiller et attendre (dans le cas des lymphomes indolents, en grande partie asymptomatiques)
Chimiothérapie
Radiothérapie (le plus souvent en cas de maladie à un stade limité et parfois en cas de maladie à un stade avancé)
Immunothérapie (p. ex., anticorps monoclonaux ou conjugués anticorps-médicaments ciblant CD20, CD19 ou CD79; anticorps bispécifiques ciblant CD20 et CD3; ou cellules CAR-T (chimeric antigen receptor T)
Médicaments ciblés (p. ex., inhibiteurs de la BTK [Bruton tyrosine kinase], inhibiteurs de la PI3K [phosphoinositide 3-kinase], inhibiteurs du cereblon, inhibiteurs de l'EZH2 [enhancer of zeste homolog 2] , inhibiteurs de la XPO1 [exportine 1])
Parfois transplantation de cellules-souches hématopoïétiques (autologue ou allogénique)
Les traitements varient considérablement selon le type cellulaire. De ce fait ces traitements sont trop nombreux pour être décrits de façon détaillée. On peut définir les grandes lignes de traitement en fonction du caractère limité ou disséminé de la maladie et de la forme agressive ou indolente. Les lymphomes de Burkitt et les lymphomes cutanés à cellules T sont discutés ailleurs. En cas de lymphomes indolents et sans signe ou symptôme significatif de lymphome, une approche "veille et attente" (arrêter le traitement tout en surveillant de près) peut être utilisée.
Maladie limitée (stades I-II)
Dans le lymphome non hodgkinien indolent de stade I (rare parce que la plupart des patients ont un stade II à IV au moment du diagnostic), la radiothérapie externe peut être le seul traitement initial. La radiothérapie régionale peut permettre un contrôle à long terme et éventuellement guérir environ 40% des patients qui ont une maladie au stade I (1, 2). Le lymphome non hodgkinien indolent de stade II est le plus souvent traité comme une maladie de stade avancé.
Les lymphomes non hodgkiniens à un stade limité peuvent être traités par une association de chimiothérapie et de radiothérapie ou par une chimiothérapie seule (plus des anticorps monoclonaux anti-CD20 pour les lymphomes à cellules B).
Maladie avancée (stades II-IV)
Le lymphome non hodgkinien de stade II est traité comme un stade avancé de la maladie dans de nombreux cas. La plupart des patients qui ont tous les types de lymphome non hodgkinien qui ont une maladie de stade II à IV sont des candidats à la chimio-immunothérapie. Dans ces cas, la radiothérapie peut être utilisée pour limiter le nombre de cycles de chimio-immunothérapie ou pour fournir un traitement localisé pour réduire les sites résiduels massifs de la maladie.
Le traitement des lymphomes de bas grade (indolents) est très variable. Les lymphomes indolents étant très traitables mais non curables, le traitement peut ne pas être recommandé initialement chez les patients qui ne présentent pas de symptômes. Certains patients qui n'ont pas de symptômes reçoivent une immunothérapie anti-CD20 en utilisant le rituximab seul. Cette stratégie peut retarder la nécessité d'introduire une chimiothérapie myélosuppressive, mais il n'a pas été démontré que l'immunothérapie précoce seule avait un impact sur la survie globale. Les patients présentant des symptômes ou ceux qui ont une maladie volumineuse qui mettent en danger les organes vitaux sont traités par chimio-immunothérapie. Dans certains cas (p. ex., chimio-réfractaires avec atteinte limitée de la moelle osseuse), l'anticorps anti-CD20 radiomarqué peut être utilisé pour cibler le rayonnement sur la cellule tumorale avec potentiellement moins d'effets sur les organes normaux proches.
Dans le cas des lymphomes B agressif (p. ex., diffus à grands lymphocytes B), le traitement standard est l'association rituximab plus cyclophosphamide, hydroxydaunorubicine (doxorubicine), vincristine, et prednisone (R-CHOP). Une réponse complète avec régression de la maladie est attendue dans 70-80% des cas, avec un taux de guérison global d'environ 60-70% (3). Ces résultats varient considérablement selon l'International Prognostic Index (IPI) pour le score du lymphome diffus à grandes cellules B. Les patients exempts de maladie à ≥ 24 mois après le diagnostic ont une espérance de vie similaire à celle de la population appariée selon l'âge et le sexe. Ce facteur clé peut guider les stratégies de suivi de cette population de patients. Les patients peuvent tirer profit de l'adjonction du conjugué anticorps-médicament polatuzumab vedotin, un conjugué anticorps-médicament dirigé contre CD79b au R-CHOP comme substitut à la vincristine (3).
L'approche dans le lymphome non hodgkinien périphérique à lymphocytes T et le lymphome primitif du système nerveux central est différente. Chez ces patients, une greffe de cellules souches autologues est proposée aux répondeurs initiaux avant que la rechute se produise dans le but d'améliorer la probabilité de guérison. Dans la transplantation de cellules souches autologues, les cellules souches sont obtenues chez le patient par leucophérèse du sang périphérique et sont transfusées chez le patient après une chimiothérapie à haute dose. De même, chez certains patients jeunes atteints de lymphome à cellules du manteau qui ont répondu au traitement initial, une greffe de cellules souches autologues peut être effectuée pour prolonger la rémission.
Rechute de lymphome
Les patients qui ont un lymphome non hodgkinien agressif non en rémission à la fin du traitement ou qui rechutent sont traités par des protocoles de chimiothérapie de deuxième ligne suivis d'une autogreffe de cellules souches s'ils sont relativement jeunes et en bonne santé. Chez certains patients à risque très élevé de rechute ainsi que chez ceux chez qui la greffe autologue n'est pas réalisable ou a déjà échoué, les cellules souches d'un frère ou d'un donneur non apparenté (greffes allogéniques) peuvent être efficaces. En général, plus le patient est âgé, moins une greffe allogénique sera proposée, car les patients âgés présentent des taux plus élevés de complications de la transplantation.
Les patients qui ont un lymphome diffus à grandes cellules B dont le lymphome est persistant ou en progression au cours des 12 mois suivant l'achèvement de l'immunochimiothérapie de première ligne ainsi que ceux qui ont un lymphome persistant malgré au moins 2 lignes de traitement antérieures peuvent être candidats au traitement par CAR-T (chimeric antigen receptor T cells). Les lymphocytes CAR T sont des lymphocytes T (le plus souvent des lymphocytes T autologues) qui ont été génétiquement modifiés pour reconnaître un antigène tumoral (p. ex., CD19). Après la perfusion, ils s'activent et subissent une expansion. Environ 60% des patients qui reçoivent des cellules CAR T comme traitement de seconde ligne et environ 30% des patients qui reçoivent un traitement CAR T comme traitement ultérieur obtiennent une réponse durable avec ce traitement (4, 5, 6, 7, 8).
Les patients non éligibles à la transplantation de cellules souches ou aux cellules CAR T, ou pour lesquels ces traitements ont échoué, peuvent recevoir un traitement par diverses thérapies, principalement pour des soins palliatifs. Ces thérapies varient considérablement et changent constamment à mesure que de nouveaux traitements sont développés.
Dans les lymphomes indolents, les patients peuvent être pris en charge au moyen d'une grande variété de stratégies en fonction des
Facteurs liés au lymphome (p. ex., histopathologie, stade, caractéristiques moléculaires et immunologiques)
Facteurs liés au patient (p. ex., âge, comorbidités)
Le type et la réponse à un traitement antérieur.
De nombreux médicaments utilisés pour le traitement de première intention peuvent être administrés aux patients en rechute. Dans certains cas, le même traitement peut être répété s'il était précédemment efficace et bien toléré. La chimiothérapie d'association à haute dose avec autogreffe de cellules souches est parfois utilisée en cas de biologie du lymphome à haut risque (y compris une mauvaise réponse à la chimiothérapie), et bien que la guérison reste peu probable, la rémission peut être de meilleure qualité que celle du traitement palliatif seul. La transplantation allogénique d'intensité réduite est une option potentiellement curative chez certains patients qui présentent un lymphome indolent. La mortalité des patients qui subissent une transplantation myéloablative a diminué de manière très importante.
Complications du traitement
Une complication immédiate de la plupart des thérapies est l'infection qui se produit pendant les périodes de neutropénie. Bien que l'utilisation de facteurs de croissance qui stimulent la production de globules blancs soit utile, l'infection continue de constituer un problème.
Les effets secondaires gastro-intestinaux de la chimiothérapie peuvent être largement soulagés ou évités par les antiémétiques et les programmes intestinaux.
Les patients recevant des anthracyclines sont à risque de cardiomyopathie et/ou d'arythmies.
Après un traitement réussi, les patients doivent être adressés à une clinique de survie au cancer pour un plan de soins qui peut être mis en œuvre par l'équipe de soins généralistes du patient. Ce programme est adapté aux comorbidités du patient et aux risques spécifiques du traitement reçu.
La chimiothérapie et la radiothérapie ont des complications tardives. Au cours des 10 premières années après le traitement, il existe un risque de myélodysplasie ou de leucémie aiguë en raison de lésions de la moelle osseuse dues à certains agents de chimiothérapie. Après 10 ans, le risque de cancers secondaires augmente surtout chez les patients qui ont reçu une radiothérapie thoracique.
Références pour le traitement
1. Lo AC, Campbell BA, Pickles T, et al. Long-term outcomes for patients with limited-stage follicular lymphoma: update of a population-based study. Blood 2020;136(8):1006-1010. doi:10.1182/blood.2019004588
2. Wilder RB, Jones D, Tucker SL, et al. Long-term results with radiotherapy for Stage I-II follicular lymphomas. Int J Radiat Oncol Biol Phys 2001;51(5):1219-1227. doi:10.1016/s0360-3016(01)01747-3
3. Tilly H, Morschhauser F, Sehn LH, et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N Engl J Med 2022;386(4):351-363. doi:10.1056/NEJMoa2115304
4. Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 2020;396(10254):839-852. doi:10.1016/S0140-6736(20)31366-0
5. Abramson JS, Solomon SR, Arnason J, et al. Lisocabtagene maraleucel as second-line therapy for large B-cell lymphoma: primary analysis of the phase 3 TRANSFORM study. Blood 2023;141(14):1675-1684. doi:10.1182/blood.2022018730
5. Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med 2022;386(7):640-654. doi:10.1056/NEJMoa2116133
7. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017;377(26):2531-2544. doi:10.1056/NEJMoa1707447
8. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 2019;380(1):45-56. doi:10.1056/NEJMoa1804980
Pronostic des lymphomes non hodgkiniens
Le pronostic varie selon le type et le stade du lymphome et les facteurs individuels du patient. En général, les patients qui ont un lymphome à lymphocytes T périphériques ou NK (natural killer)/T ont un pronostic plus sombre que ceux qui ont un lymphome non hodgkinien à lymphocytes B. Le pronostic de chacun des sous-types de lymphomes non hodgkiniens est lié aux différences de la biologie cellulaire tumorale.
Le système de cotation pronostique le plus couramment utilisé est l'International Prognostic Index (IPI) for diffuse large B-cell lymphoma. Cependant, le score IPI n'est utilisé que dans le lymphome diffus à grandes cellules B. Il existe également des systèmes de notation du lymphome folliculaire (FLIPI) et du lymphome à cellules du manteau (MIPI). Des calculatrices en ligne permettent d'estimer le pronostic dans d'autres types de lymphome non hodgkinien également.
L'IPI (International Prognostic Index) prend en compte 5 facteurs de risque:
Âge > 60 ans
Un mauvais "performance status" (pouvant être évalué en utilisant le système de l'Eastern Cooperative Oncology Group tool)
Taux élevé de lactate déshydrogénase (LDH)
> 1 site extra-ganglionnaire
Une maladie au stade III ou IV
La survie diminue avec l'augmentation du nombre de facteurs de risque. Les patients n'ayant aucun de ces facteurs de risque ont un taux de guérison très élevé. Le score initial de l'IPI utilise les 5 facteurs comme variables discrètes (p. ex., un âge de plus de 60 ans ou de moins de 60 ans). Une modification, la Diffuse Large B-cell Lymphoma Prognosis (IPI24), qui calcule la probabilité d'être indemne de maladie à 24 mois après le diagnostic, intègre les facteurs ci-dessus comme variables continues et intègre également le nombre absolu de lymphocytes.
Points clés
Les lymphomes non hodgkiniens sont un groupe de cancers connexes impliquant les lymphocytes; ils varient de manière significative en matière de taux de croissance et de réponse au traitement.
Généralement, la maladie avait déjà diffusé au moment du diagnostic.
Les tests moléculaires et génétiques sont essentiels pour le diagnostic et la prise en charge.
Une maladie indolente limitée peut être traitée par radiothérapie.
Traiter la maladie plus avancée (indolente ou agressive) par l'immunothérapie, la chimiothérapie, la greffe de cellules souches hématopoïétiques ou une association en fonction du type et du stade du lymphome non hodgkinien.
Plus d'information
Ce qui suit est une ressource en anglais qui fournit des informations aux médecins et un soutien et des informations aux patients. LE MANUEL n'est pas responsable du contenu de cette ressource.
Leukemia & Lymphoma Society: Resources for Healthcare Professionals: fournit des ressources de formation pour les professionnels de santé ainsi que des informations pour l'orientation des patients







