



Lupus nephritis is glomerulonephritis caused by systemic lupus erythematosus (SLE). Clinical findings include hematuria, nephrotic-range proteinuria (≥ 3 g/day), and, in advanced stages, azotemia. Diagnosis is based on renal biopsy. Treatment is of the underlying disorder and usually involves corticosteroids and other immunosuppressant medications.
Lupus nephritis is present in 20 to 60% of patients with SLE (1). In the United States, it disproportionally affects minorities, particularly African Americans (2).
General references
1. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int 2021;100(4S):S1-S276. doi:10.1016/j.kint.2021.05.021
2. Portalatin GM, Gebreselassie SK, Bobart SA. Lupus nephritis - An update on disparities affecting African Americans. J Natl Med Assoc 2022;114(3S2):S34-S42. doi:10.1016/j.jnma.2022.05.005
Pathophysiology of Lupus Nephritis
Pathophysiology involves immune complex deposition with development of glomerulonephritis. The immune complexes consist of
Nuclear antigens (especially DNA)
High-affinity complement-fixing IgG antinuclear antibodies
Antibodies to DNA
Subendothelial, intramembranous, subepithelial, or mesangial deposits are characteristic. Wherever immune complexes are deposited, immunofluorescence staining is positive for complement and for IgG, IgA, and IgM in varying proportions. Epithelial cells may proliferate, forming crescents.
Classification of lupus nephritis is based on histologic findings (see table ).
Antiphospholipid syndrome nephropathy
This syndrome may occur with or without lupus nephritis in up to one-third of patients with SLE. The syndrome occurs in the absence of any other autoimmune process in approximately 30% of affected patients (1). In antiphospholipid syndrome, circulating lupus anticoagulant causes microthrombi, endothelial damage, and cortical ischemic atrophy. Antiphospholipid syndrome nephropathy increases a patient’s risk of hypertension and chronic kidney disease or kidney failure compared with lupus nephritis alone.
Pathophysiology reference
1. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int 2021;100(4S):S1-S276. doi:10.1016/j.kint.2021.05.021
Symptoms and Signs of Lupus Nephritis
The most prominent symptoms and signs are those of SLE; patients who present with kidney disease may have edema, hypertension, or a combination.
Classification of Lupus Nephritis
Class | Description | Histologic Findings* | Clinical Findings | Renal Prognosis |
|---|---|---|---|---|
I | Minimal mesangial | Normal (although immune complexes are sometimes visible using immunofluorescence or electron microscopy) | None | Excellent |
II | Mesangial proliferative | Immune complexes in the mesangium only and mesangial hypercellularity | Possibly microscopic hematuria, proteinuria, or both | Excellent |
III | Focal | Endocapillary and extracapillary cellular hypercellularity and inflammation in < 50% of glomeruli, usually in a segmental distribution | Usually hematuria and proteinuria Possibly hypertension, nephrotic syndrome, and elevated serum creatinine | Variable |
IV | Diffuse† | Endocapillary and extracapillary cellular hypercellularity and inflammation in > 50% of glomeruli | Usually hematuria and proteinuria Frequently hypertension, nephrotic syndrome, and elevated serum creatinine | Variable |
V | Membranous | Thickening of the glomerular basement membrane with subepithelial and intramembranous immune complex deposition | Usually nephrotic syndrome Sometimes microscopic hematuria or hypertension Serum creatinine usually normal or slightly elevated | Poorly defined |
VI | Advanced sclerosing | Sclerosis of > 90% of glomerular capillaries | Bland urinary sediment and kidney failure or slowly increasing serum creatinine | Poor |
* Using light microscopy. | ||||
† Most common form. | ||||
Data from Weening JJ, D'Agati VD, Schwartz MM, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited [published correction appears in Kidney Int. 2004 Mar;65(3):1132]. Kidney Int 2004;65(2):521-530. doi:10.1111/j.1523-1755.2004.00443.x and Bajema IM, Wilhelmus S, Alpers CE, et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. 2018;93(4):789-796. doi:10.1016/j.kint.2017.11.023 | ||||
Diagnosis of Lupus Nephritis
Urinalysis and serum creatinine (all patients with SLE)
Diagnosis is suspected in all patients with SLE, particularly in patients who have proteinuria, microscopic hematuria, red blood cell (RBC) casts, or hypertension. Diagnosis is also suspected in patients with unexplained hypertension, elevated serum creatinine levels, or abnormalities on urinalysis who have clinical features suggesting SLE.
Initially, urinalysis is performed and serum creatinine as well as spot protein to creatine ratio, anti–double-stranded-DNA (anti-dsDNA), and complement (C3 and C4) levels are measured (1). Elevated anti-dsDNA antibody titers and low complement levels often indicate active lupus nephritis and support the diagnosis.
If the aforementioned studies are abnormal, particularly in the cases of declining GFR or confirmed proteinuria, renal biopsy is usually performed to confirm the diagnosis and classify the disorder histologically (1). Histologic classification helps determine prognosis and direct treatment (see table ).
Some of the histologic subtypes are similar to other glomerulopathies; eg, membranous lupus nephritis is histologically similar to idiopathic membranous nephropathy and diffuse proliferative lupus nephritis is histologically similar to type I membranoproliferative glomerulonephritis. Overlap between these categories is substantial, and patients may progress from one class to another.

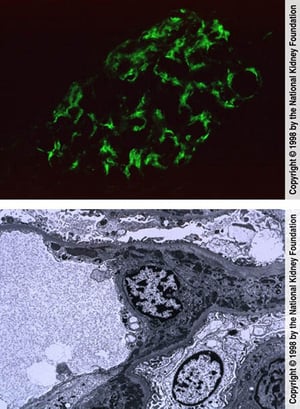
The top image shows mesangial deposition of IgM (immunofluorescence with anti-IgM, original magnification ×400). In the bottom image, mesangial dense immune complex deposits and reticular aggregates within endothelial cell cytoplasm are seen on transmission electron microscopy (×8000).
The top image shows mesangial deposition of IgM (immunofluorescence with anti-IgM, original magnification ×400). In the
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).

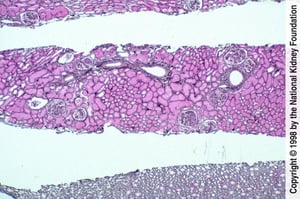
Focal lupus nephritis is characterized by endocapillary proliferation caused by immune complex deposits involving < 50% of glomeruli in a biopsy specimen.
Focal lupus nephritis is characterized by endocapillary proliferation caused by immune complex deposits involving <
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
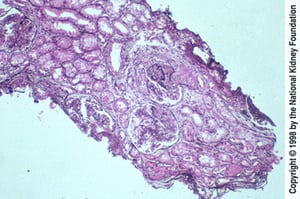
Diffuse lupus nephritis is characterized by endocapillary proliferation caused by immune complex deposits involving > 50% of glomeruli in a biopsy specimen.
Diffuse lupus nephritis is characterized by endocapillary proliferation caused by immune complex deposits involving >
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
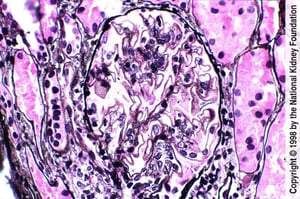
Diffuse holes, small spikes, and mild mesangial expansion with small areas of pink staining indicate mesangial immune complex deposits (Jones silver stain, ×400).
Diffuse holes, small spikes, and mild mesangial expansion with small areas of pink staining indicate mesangial immune c
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
The top image shows mesangial deposition of IgM (immunofluorescence with anti-IgM, original magnification ×400). In the bottom image, mesangial dense immune complex deposits and reticular aggregates within endothelial cell cytoplasm are seen on transmission electron microscopy (×8000).
The top image shows mesangial deposition of IgM (immunofluorescence with anti-IgM, original magnification ×400). In the
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Focal lupus nephritis is characterized by endocapillary proliferation caused by immune complex deposits involving < 50% of glomeruli in a biopsy specimen.
Focal lupus nephritis is characterized by endocapillary proliferation caused by immune complex deposits involving <
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Diffuse lupus nephritis is characterized by endocapillary proliferation caused by immune complex deposits involving > 50% of glomeruli in a biopsy specimen.
Diffuse lupus nephritis is characterized by endocapillary proliferation caused by immune complex deposits involving >
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Diffuse holes, small spikes, and mild mesangial expansion with small areas of pink staining indicate mesangial immune complex deposits (Jones silver stain, ×400).
Diffuse holes, small spikes, and mild mesangial expansion with small areas of pink staining indicate mesangial immune c
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Kidney function and SLE activity should be monitored regularly. A rising serum creatinine level reflects deteriorating kidney function, while falling serum C3 and C4 levels or a rising anti-dsDNA antibody titer suggests increased disease activity.
Diagnosis reference
1. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int 2021;100(4S):S1-S276. doi:10.1016/j.kint.2021.05.021
Treatment of Lupus Nephritis
Angiotensin inhibition for hypertension or proteinuria
Corticosteroids plus either mycophenolate or cyclophosphamide for active, potentially reversible nephritis
Sometimes other immunosuppressive agents (eg, belimumab, calcineurin inhibitors) are added to the initial regimen
Kidney transplantation for patients with kidney failure
Angiotensin inhibition with an angiotensin-converting enzyme (ACE) inhibitor or angiotensin II receptor blocker (ARB) is indicated for patients with even mild hypertension (eg, blood pressure [BP] > 130/80 mm Hg) or proteinuria. Also, dyslipidemia and risk factors for atherosclerosis should be treated aggressively.
Immunosuppression
Treatment is guided by the histologic classification of the lupus nephritis, the degree of disease activity and chronicity, and the presence of concomitant kidney disorders.
Activity is estimated by the activity score as well as clinical criteria (eg, urine sediment, increasing urine protein, increasing serum creatinine). Many experts believe that a mild to moderate chronicity score, because it suggests reversibility, should provoke more aggressive therapy than a more severe chronicity score. Nephritis with the potential for deterioration and for reversibility is usually class III or IV; it is unclear whether class V nephritis warrants aggressive treatment.
The activity score describes the degree of inflammation. The score is based on cellular proliferation, fibrinoid necrosis, cellular crescents, hyaline thrombi, wire loop lesions, glomerular leukocyte infiltration, and interstitial mononuclear cell infiltration. The activity score is less well correlated with disease progression and is used, rather, to help identify active nephritis.
The chronicity index describes the degree of scarring. It is based on presence of glomerular sclerosis, fibrous crescents, tubular atrophy, and interstitial fibrosis. The chronicity index predicts progression of lupus nephritis to kidney failure. A mild to moderate chronicity score suggests at least partially reversible disease, whereas more severe chronicity scores may indicate irreversible disease.
Treatment for proliferative lupus nephritis combines corticosteroids with other immunosuppressants (1, 2).
Induction therapy for focal or diffuse lupus nephritis usually consists of corticosteroids in combination with either mycophenolate or intravenous cyclophosphamide (see table ). Prednisone is typically begun at 60 to 80 mg orally once a day and tapered according to response over 6 to 12 months. There is no consensus regarding the optimal corticosteroid dosing regimen. Lower starting doses of prednisone followed by faster tapers may also be used. Relapses are usually treated with increasing doses of prednisone and sometimes a change in the adjunctive immunosuppressive agent. Cyclophosphamide and mycophenolate are equally efficacious, although systemic toxicity may be less with mycophenolate than with cyclophosphamide (3). Reasonable alternatives for induction therapy include mycophenolate in combination with either a calcineurin inhibitor (tacrolimus or voclosporin) or belimumab (4, 5).
After an appropriate renal response has been achieved with induction therapy, immunosuppressive therapy is continued for at least 2 years and often longer. When cyclophosphamide is used as initial therapy, patients are switched to mycophenolate for maintenance therapy to limit toxicity from cyclophosphamide. Mycophenolate is also preferred over azathioprine for maintenance therapy because of higher rates of relapse observed with azathioprine use. However, azathioprine is preferred for patients who want to become pregnant. Azathioprine is also a reasonable option for patients who are intolerant to mycophenolate or have limited access due to cost. Low-dose prednisone is continued in most patients and titrated based on disease activity.
For patients with pure lupus membranous nephropathy, there is a lack of consensus regarding the role of immunosuppressive therapy, and some experts limit its use depending on the degree of proteinuria and findings on kidney biopsy. However, patients who have concurrent lupus membranous nephropathy and focal or diffuse lupus nephritis are treated using the same approach as for focal or diffuse lupus nephritis alone.
Other treatments
Anticoagulation is of theoretical benefit for patients with antiphospholipid syndrome nephropathy, but the value of such treatment has not been established. However, patients with antiphospholipid syndrome and a definite thrombotic event should be anticoagulated in accordance with preferred agents for antiphospholipid syndrome.
Kidney transplantation is an option for patients with kidney failure due to lupus nephritis. Recurrent disease in the graft is uncommon (< 5%), but risk may be increased in Black people, females, and younger patients (6).
Treatment references
1. Fanouriakis A, Kostopoulou M, Cheema K, et al: 2019 Update of the Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann Rheum Dis 9(6):713-723, 2020. doi: 10.1136/annrheumdis-2020-216924
2. Kidney Disease: Improving Global Outcomes (KDIGO) Lupus Nephritis Work Group: KDIGO 2024 Clinical Practice Guideline for the management of LUPUS NEPHRITIS. Kidney Int 105(1S):S1–S69, 2024. doi:10.1016/j.kint.2023.09.002
3. Tunnicliffe DJ, Palmer SC, Henderson L, et al: Cochrane Database Syst Rev 6(6):CD002922, 2018. doi: 10.1002/14651858.CD002922.pub4
4. Rovin BH, Onno Teng YK, Ginzler EM, et al: Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 397(10289):2070-2080, 2021. doi: 10.1016/S0140-6736(21)00578-X
5. Rovin BH, Furie R, Onno Teng YK, et al: A secondary analysis of the Belimumab International Study in Lupus Nephritis trial examined effects of belimumab on kidney outcomes and preservation of kidney function in patients with lupus nephritis. Kidney Int 101(2):403-413, 2022. doi: 10.1016/j.kint.2021.08.027
6. Contreras G, Mattiazzi A, Guerra G, et al: Recurrence of lupus nephritis after kidney transplantation. J Am Soc Nephrol 21(7):1200–1207, 2010. doi:10.1681/ASN.2009101093
Prognosis for Lupus Nephritis
Class of nephritis influences renal prognosis (see table ), as do other renal histologic features, baseline kidney function, and race (1).
Patients with lupus nephritis are at high risk of cancers, primarily B-cell lymphomas. Risk of atherosclerotic complications (eg, coronary artery disease, ischemic stroke) is also high because of frequent vasculitis, hypertension, dyslipidemia, and use of corticosteroids.
Prognosis reference
1. Portalatin GM, Gebreselassie SK, Bobart SA. Lupus nephritis - An update on disparities affecting African Americans. J Natl Med Assoc 2022;114(3S2):S34-S42. doi:10.1016/j.jnma.2022.05.005
Key Points
Nephritis is diagnosed in up to 60% of patients with SLE and is likely present in more.
Obtain urinalysis and measure serum creatinine in all patients with SLE and renal biopsy if an unexplained abnormality is found in either, particularly in the presence of low C3 and C4 and elevated dsDNA.
Initiate angiotensin inhibition for even mild hypertension and treat atherosclerotic risk factors aggressively.
Treat active, potentially reversible nephritis with corticosteroids plus mycophenolate and/or cyclophosphamide.
Continue maintenance therapy for at least 2 years, which typically includes mycophenolate and low-dose prednisone.
Drug Information for the Topic





