







Le lymphome d'Hodgkin est une prolifération maligne, localisée ou disséminée, de cellules du système lymphoréticulaire, touchant principalement les ganglions, la rate, le foie et la moelle osseuse. La symptomatologie comprend typiquement la présence d'une adénopathie indolore, parfois associée à une fièvre, des sueurs nocturnes, une perte de poids involontaire, un prurit, une splénomégalie et une hépatomégalie. Le diagnostic repose sur la biopsie ganglionnaire. Le traitement est curatif dans la plupart des cas et consiste en une chimiothérapie avec ou sans autres modalités de traitement, dont des conjugués anticorps-médicaments, une immunothérapie et une radiothérapie.
(Voir aussi Revue générale des lymphomes.)
Aux États-Unis, on estime qu'en 2023, environ 8830 nouveaux cas de lymphome de Hodgkin auront été diagnostiqués et qu'environ 900 personnes seront décédées de la maladie (1). Le ratio homme/femme est d'environ 1,2:1. Le lymphome d'Hodgkin, rare avant 10 ans est le plus fréquent entre 15 et 40 ans; avec un 2e pic > 60 ans.
Référence générale
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73(1):17-48. doi:10.3322/caac.21763
Physiopathologie du lymphome de Hodgkin
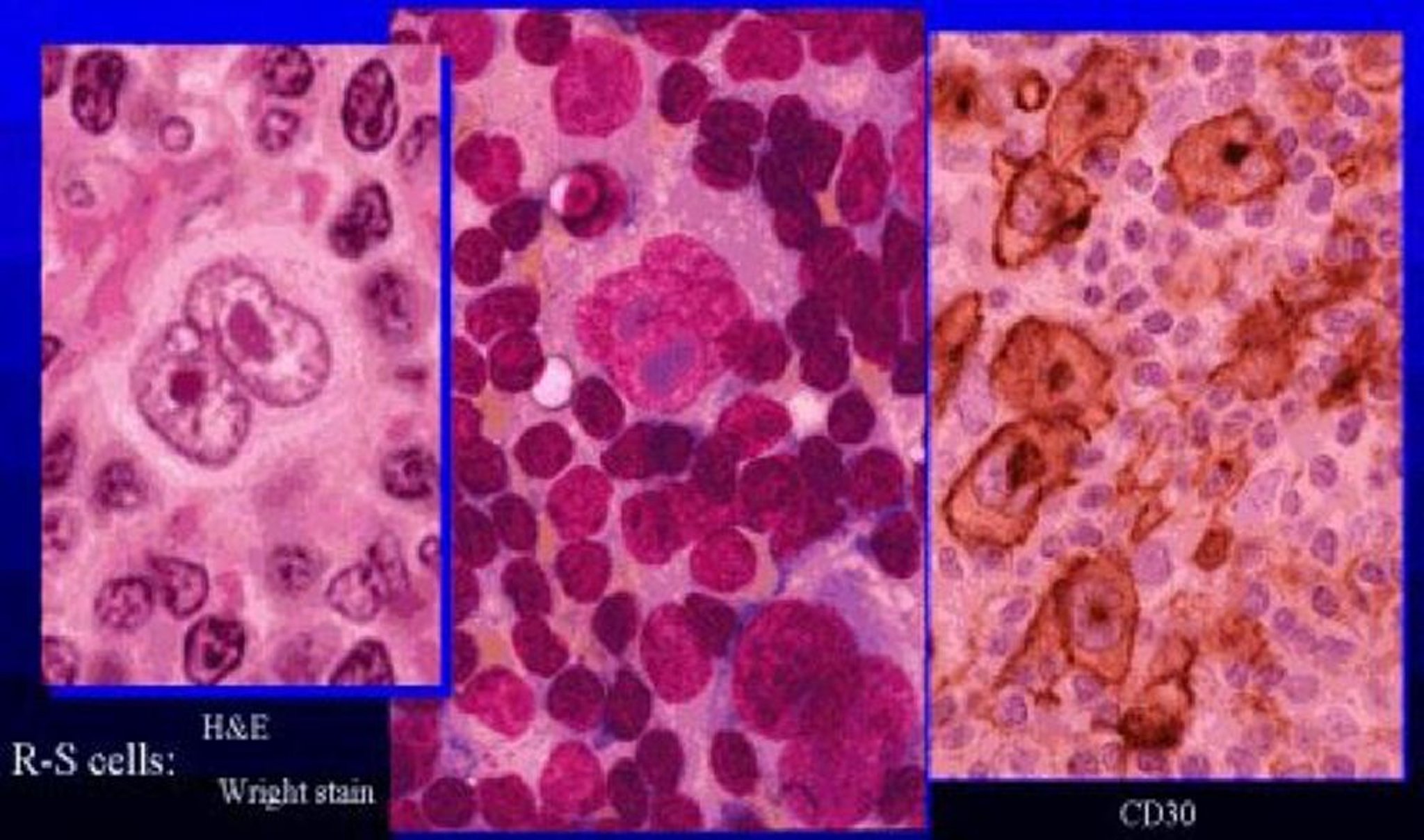
Le lymphome d'Hodgkin résulte de la transformation clonale de cellules lymphoïdes appartenant à la lignée B, donnant naissance aux cellules binucléées de Reed-Sternberg, pathognomoniques de la maladie.
La cause est inconnue, mais la prédisposition génétique (p. ex., antécédents familiaux) et les associations environnementales jouent un rôle. Les associations environnementales liées au lymphome de Hodgkin comprennent les antécédents de traitement par la phénytoïne, la radiothérapie ou la chimiothérapie et l'infection par le Virus Epstein-Barr (EBV) ou le VIH. Le risque est légèrement augmenté en cas de
Certains types d'immunosuppression (p. ex., les patients transplantés qui prennent des immunosuppresseurs)
Syndrome d'immunodéficience congénitale (p. ex., ataxie-télangiectasie, syndrome de Klinefelter, syndrome de Chédiak-Higashi, syndrome de Wiskott-Aldrich)
Certaines maladies auto-immunes (polyarthrite rhumatoïde, maladie cœliaque, syndrome de Sjögren, lupus érythémateux disséminé)
La plupart des patients développent par ailleurs un déficit lentement progressif de l'immunité cellulaire (fonction lymphocytaire T), qui contribue aux infections bactériennes à germe banal et aux infections mycosiques, virales ou parasitaires inhabituelles, observées aux stades avancés de la maladie. L'immunité humorale (production d'anticorps) est également altérée à ces stades avancés. La mort peut résulter d'une infection ou d'une maladie évolutive.
Symptomatologie du lymphome de Hodgkin
La plupart des patients qui ont un lymphome de Hodgkin ont à la présentation une adénopathie cervicale indolore. Bien que le mécanisme n'en soit pas clair, une douleur peut rarement survenir dans le territoire atteint immédiatement après l'absorption de boissons alcoolisées, orientant parfois précocement le diagnostic.
D'autres manifestations apparaissent au fur et à mesure de la dissémination de la maladie dans le système réticulo-endothélial, généralement par contiguïté. Un prurit intense réfractaire aux thérapies habituelles peut survenir précocement.
Les symptômes généraux associent une fièvre, des sueurs nocturnes et une perte d'appétit entraînant une perte de poids involontaire (> 10% du poids du corps au cours des 6 derniers mois) qui sont appelés "symptômes B." Les symptômes B sont importants pour le pronostic et la classification par stade, car ils peuvent traduire la présence d'adénopathies profondes (médiastinales ou rétropéritonéales), d'une atteinte viscérale (foie) ou médullaire. Une splénomégalie est fréquente; une hépatomégalie est inhabituelle. Une fièvre de Pel-Ebstein (quelques jours de fièvre élevée alternant régulièrement avec quelques jours à quelques semaines de température normale ou en dessous de la normale) est parfois observée. Une cachexie apparaît fréquemment avec la progression de la maladie.
Bone involvement is often asymptomatic but may cause vertebral osteoblastic lesions (ivory vertebrae) and, rarely, pain with osteolytic lesions and compression fractures.
Les lésions intracrâniennes, gastriques et cutanées sont rares et, lorsqu'elles sont présentes, elles peuvent suggérer un lymphome de Hodgkin non contrôlé associé au VIH.
Les masses tumorales peuvent être compressives et de ce fait entraîner des symptômes comme
Un ictère secondaire à une obstruction des voies biliaires intra- ou extrahépatiques
Œdème localisé (lymphœdème) secondaire à une obstruction lymphatique par la tumeur
Une dyspnée sévère avec wheezing secondaire à une compression trachéobronchique en raison d'une maladie médiastinale
Dyspnée, toux et/ou gêne thoracique dues à l'infiltration du parenchyme pulmonaire, qui peut simuler une consolidation ou une bronchopneumonie lobaires
Un envahissement épidural comprimant la moelle épinière peut entraîner une paraplégie.
Un syndrome de Claude Bernard-Horner et une paralysie laryngée peuvent résulter d'une compression cervicale du nerf sympathique ou récurrent par des ganglions lymphatiques tumoraux.
La compression des racines nerveuses entraîne des douleurs névralgiques.
Diagnostic du lymphome de Hodgkin
Biopsie de ganglion lymphatique
FDG-PET/TDM thoracique et abdomino-pelvienne pour la classification par stades
IRM si des symptômes neurologiques sont présents
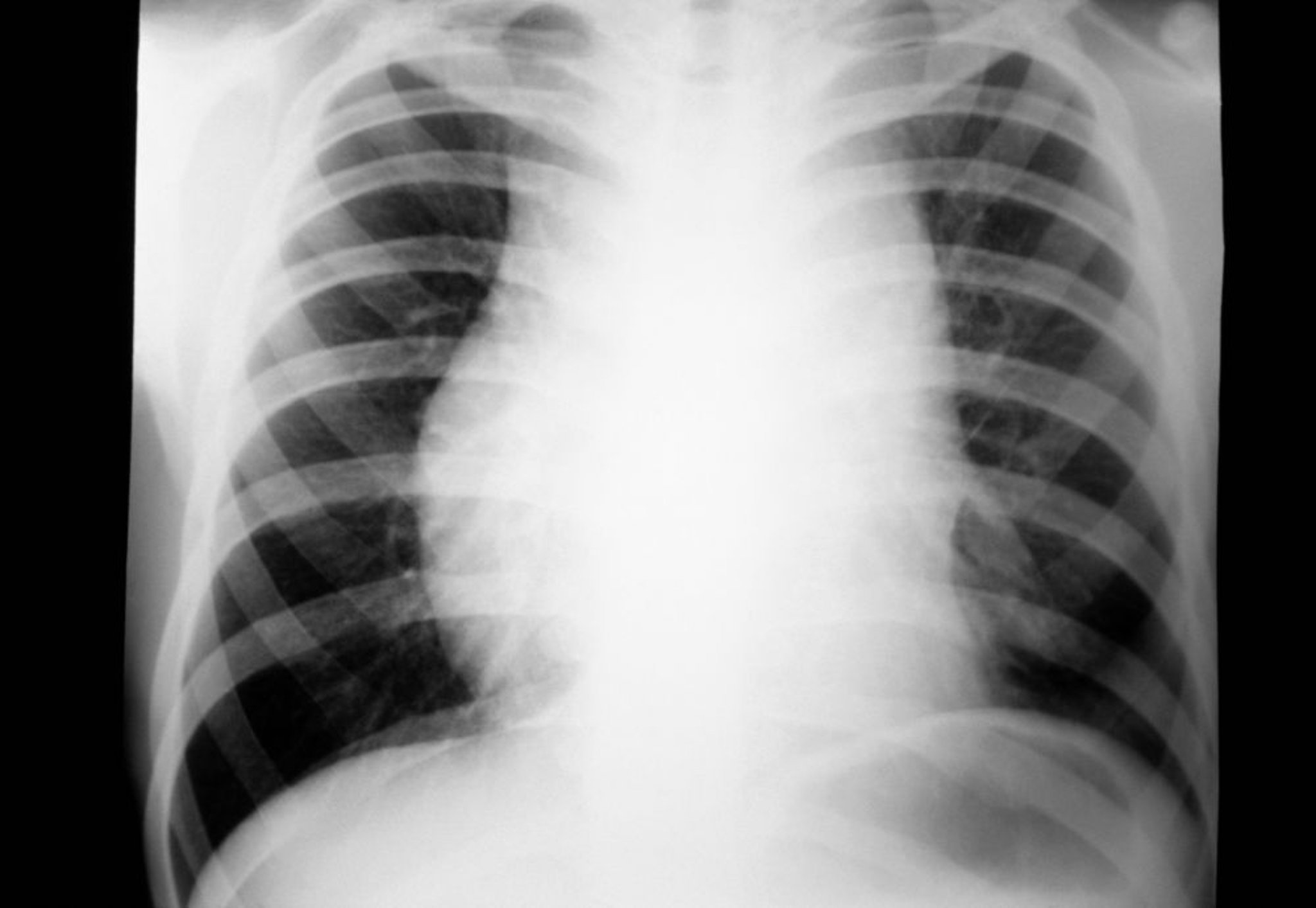
Le diagnostic de lymphome d'Hodgkin est habituellement évoqué en cas d'adénopathie périphérique indolore ou d'adénopathie médiastinale découverte à l'occasion d'un examen clinique ou d'une rx thorax de routine (1).
Une lymphadénopathie similaire peut résulter d'infections virales telles que: la mononucléose infectieuse (EBV) ou l'infection par le cytomégalovirus (CMV), la toxoplasmose, le lymphome non hodgkinien, ou la leucémie. Des signes similaires à la rx thorax peuvent résulter d'un cancer du poumon, d'une sarcoïdose ou d'une tuberculose.
(Le bilan d'une masse médiastinale est traité ailleurs.)

DR P. MARAZZI/SCIENCE PHOTO LIBRARY
La radiographie thoracique ou les anomalies de l'examen clinique doivent être confirmées par TDM ou PET (positron emission tomography) scan du thorax afin de choisir la procédure de biopsie la plus efficace. Si seuls les ganglions médiastinaux sont augmentés de volume, une médiastinoscopie, une thoracoscopie assistée par vidéo ou une intervention de Chamberlain (thoracostomie antérieure gauche limitée, permettant la biopsie de ganglions médiastinaux inaccessibles par médiastinoscopie cervicale) sont indiquées. Une biopsie à l'aiguille guidée par TDM peut également être envisagée; l'aspiration à l'aiguille fine est souvent inappropriée pour le diagnostic du lymphome de Hodgkin.
La biopsie montre des cellules de Reed-Sternberg (grandes cellules binucléées) au sein d'un infiltrat cellulaire hétérogène caractéristique composé d'histiocytes, de lymphocytes, de monocytes, de plasmocytes et d'éosinophiles. Le lymphome de Hodgkin classique a 4 sous-types histopathologiques (2) (voir tableau Sous-types histopathologiques des lymphomes d'Hodgkin):
Sclérose nodulaire: tissu fibreux dense découpant le parenchyme ganglionnaire en nodules de tissu hodgkinien
Cellularité mixte : a sein d'une population cellulaire polymorphe, présence d'un nombre modéré de cellules de Reed-Sternberg
Riche en lymphocytes: peu de cellules de Reed-Sternberg mais beaucoup de cellules B
A déplétion lymphocytaire: nombreuses cellules de Reed-Sternberg plus fibrose extensive
Le lymphome hodgkinien nodulaire à prédominance lymphocytaire a été reclassé en lymphome B non hodgkinien par l'International Consensus Classification et est appelé lymphome nodulaire à lymphocytes B prédominant (3).

Courtoisie de la FDA.
Une NFS, une vitesse de sédimentation érythrocytaire (VS), un dosage des LDH (lactate déshydrogénase), un bilan rénal et hépatique sont généralement pratiqués. Les tests peuvent être anormaux mais ne sont pas diagnostiques.
La NFS peut révéler une légère hyperleucocytose à polynucléaires neutrophiles. Une lymphopénie peut apparaître précocement et constitue un facteur pronostique défavorable. L'éosinophilie est fréquente et une thrombocytose peut également être présente. Une anémie, souvent microcytaire, se développe habituellement aux stades avancés de la maladie. Dans l'anémie avancée, la réutilisation défectueuse du fer est caractérisée par une faible teneur en fer sérique, une faible capacité de fixation du fer, une ferritine sérique élevée et une augmentation de la teneur en fer de la moelle osseuse. La pancytopénie est parfois provoquée par une invasion de la moelle osseuse, le plus souvent dans le sous-type à déplétion lymphocytaire.
Une augmentation des phosphatases alcalines sériques peut être observée, mais n'indique pas nécessairement une atteinte médullaire et/ou hépatique. L'élévation des phosphatases alcalines leucocytaires, de l'haptoglobine sérique, et des autres marqueurs inflammatoires de phase aiguë témoignent habituellement de la présence de cytokines inflammatoires dues à un lymphome de Hodgkin actif. Ces substances sont mesurées et des tests sont parfois effectués pour évaluer des symptômes non spécifiques et les résultats peuvent suggérer un lymphome de Hodgkin; ils ne sont pas pratiqués chez tous les patients atteints de lymphome. La vitesse de sédimentation érythrocytaire, un marqueur indirect de l'inflammation, est souvent ordonnée et son élévation prédit une évolution moins favorable.
Un hypersplénisme peut être observé en cas de splénomégalie importante.
Une scintigraphie PET-FDG (fluorodésoxyglucose)-/TDM du thorax, de l'abdomen et du bassin est l'imagerie de choix pour la classification par stades du lymphome de Hodgkin (voir plus loin). Les lésions osseuses sont détectées plus fréquemment par l'imagerie FDG-PET. Si la FDG-PET/TDM n'est pas disponible, une TDM du thorax, de l'abdomen et du bassin est effectué.
Une biopsie de moelle osseuse n'est habituellement effectuée que si une PET/TDM n'est pas possible et si les signes peuvent modifier la prise en charge.
D'autres examens complémentaires sont effectués en fonction des signes (p. ex., IRM en cas de symptômes de compression médullaire). D'autres tests recommandés comprennent la fraction d'éjection cardiaque si l'utilisation d'anthracyclines est prévue et les épreuves fonctionnelles respiratoires si la bléomycine est envisagée.
Classification par stades
Une fois le diagnostic établi, le stade est déterminé afin de guider le traitement. Le système de classification par stades de Lugano couramment utilisé (voir tableau Classification par stades de Lugano du lymphome hodgkinien et des lymphomes non hodgkiniens) intègre
Symptômes
Résultats de l'examen clinique
L'imagerie, dont la rx thorax, la TDM du thorax, et abdomino-pelvienne et la PET-FDG.
Parfois signes à la biopsie de moelle osseuse
La laparotomie n'est pas nécessaire à la classification par stades.
Références pour le diagnostic
1. Cheson BD, Fisher RI, Barrington SF, et al: Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J Clin Oncol 32(27):3059–3068, 2014.
2. Alaggio R, Amador C, Anagnostopoulos I, et al: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms [published correction appears in Leukemia 2023 Sep;37(9):1944-1951]. Leukemia 36(7):1720–1748, 2023. doi:10.1038/s41375-022-01620-2
3. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee [published correction appears in Blood 2023 Jan 26;141(4):437]. Blood 2022;140(11):1229-1253. doi:10.1182/blood.2022015851
Traitement du lymphome de Hodgkin
Chimiothérapie
Conjugué anticorps-médicament (p. ex., brentuximab vedotin)
Immunothérapie (p. ex., immune checkpoint inhibitors [inhibiteurs des points de contrôle immunitaires])
Radiothérapie
Parfois, transplantation de cellules-souches autologues
Le choix des modalités thérapeutiques est complexe et dépend du stade précis de la maladie. Avant le traitement et le cas échéant, les patientes doivent discuter des options pour préserver la fertilité avec leur oncologue et un spécialiste de la fertilité.
Traitement initial
Les stades limités de la maladie sont généralement traités par un nombre limité de cycle de polychimiothérapie comprenant de la doxorubicine (adriamycine), bléomycine, vinblastine et dacarbazine (ABVD) avec ou sans radiothérapie. En cas de masse médiastinale volumineuse, la chimiothérapie peut être prolongée ou de nature différente et la radiothérapie est souvent utilisée. La bléomycine est généralement évitée chez les patients > 60 ans en raison d'un risque accru de toxicité pulmonaire.
Le stade avancé de la maladie est généralement pris en charge par des protocoles basés sur l'AVD. Des options raisonnables peuvent comprendre l'ABVD, le BEACOPP (bléomycine, étoposide, doxorubicine, cyclophosphamide, vincristine, procarbazine et prednisone), l'AVD plus brentuximab védotine, et l'AVD plus nivolumab, qui est en passe de devenir la nouvelle norme de soins. Le choix du traitement de la maladie à stade avancé est individualisé et basé sur les caractéristiques du patient (p. ex., stade de la maladie, comorbidités), les toxicités médicamenteuses potentielles et les préférences du patient. Ces approches alternatives sont largement basées sur les résultats de grands essais randomisés. Dans l'essai RATHL (Response-Adapted Therapy in Advanced Hodgkin Lymphoma), les patients ont été traités par ABVD, et ceux dont la PET est négative après 2 cycles ont reçu 4 cycles supplémentaires d'AVD (pas de bléomycine), alors que ceux dont la PET est positive ont été intensifiés en BEACOPP (bléomycine, étoposide, doxorubicine, cyclophosphamide, vincristine, procarbazine et prednisone [1]). Dans l'essai ECHELON-1, les patients traités par AVD plus le conjugué anticorps-médicament anti-CD30 brentuximab vedotin ont eu des bénéfices supérieurs à ceux des patients traités par ABVD, les patients plus jeunes à risque élevé semblant en bénéficier le plus (2, 3). La prise en charge optimale des patients très âgés ou fragiles n'est pas standardisée.
Traitement ultérieur
Plusieurs protocoles de chimiothérapie de deuxième ligne sont considérés comme acceptables chez les patients qui ne sont pas guéris par le traitement de première ligne. Chez les patients qui ont une bonne réponse au traitement de seconde intention, une chimiothérapie à haute dose et une autogreffe de cellules souches doit être envisagé alors que les non-répondeurs peuvent être candidats à d'autres traitements ou à une greffe de cellules souches allogéniques.
Le brentuximab vedotin et les inhibiteurs de point de contrôle (nivolumab) et le pembrolizumab peuvent être utilisés dans le traitement des patients atteints de lymphome de Hodgkin qui ont reçu auparavant au moins 2 formes de traitement (4, 5).
Complications du traitement
La chimiothérapie, en particulier avec des médicaments tels que les agents alkylants (méchloréthamine, cyclophosphamide, procarbazine), doxorubicine et étoposide, augmente le risque de leucémie entre 3 et 10 ans après la thérapie.
La radiothérapie comporte un risque accru de tumeurs solides malignes (p. ex., du sein, gastro-intestinales, pulmonaires, thyroïdiennes, des tissus mous).
La doxorubicine ainsi que les irradiations médiastinales augmentent le risque de cardiomyopathie, d'athérosclérose coronaire et de valvulopathie.
La bléomycine peut induire des lésions pulmonaires, qui peuvent être graves et rarement mortelles.
Le brentuximab vedotin est myélosuppresseur et en particulier lorsqu'il est associé à la vinblastine, il peut induire une neuropathie périphérique durable.
Les inhibiteurs de points de contrôle immunitaire (Immune checkpoint inhibitors) sont associés à des toxicités de type immunitaire.
Surveillance après traitement
Tous les patients qui n'ont pas une PET négative à la fin du traitement d'induction doivent subir une biopsie ou être suivis attentivement par une imagerie en série; en cas de maladie résiduelle, un traitement supplémentaire est nécessaire. Une fois en rémission, les patients doivent être suivis à la recherche d'une symptomatologie de rechute pendant 5 ans. En cas de signes de rechute, définie comme une réapparition de la maladie au niveau de sites où la maladie s'est déjà manifestée ou de nouveaux sites, doivent bénéficier d'une imagerie par PET/TDM ou TDM seule. Une imagerie de routine chez les patients asymptomatiques n'est pas obligatoire. Pour un calendrier de surveillance post-traitement, voir tableau Surveillance post-thérapeutique des lymphomes d'Hodgkin.
Références pour le traitement
1. Johnson P, Federico M, Kirkwood A, et al: Adapted treatment guided by interim PET-CT scan in advanced Hodgkin's lymphoma. N Engl J Med 374(25):2419– 2429, 2016.
2. Connors JM, Jurczak W, Straus DJ, et al: Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin's lymphoma. N Engl J Med 378(4):331–344, 2018. Epub 2017 Dec 10.
3. Straus DJ, Długosz-Danecka M, Connors JM, et al: Brentuximab vedotin with chemotherapy for stage III or IV classical Hodgkin lymphoma (ECHELON-1): 5-year update of an international, open-label, randomised, phase 3 trial. Lancet Haematol 8(6):e410–e421, 2021. doi: 10.1016/S2352-3026(21)00102-2
4. Armand P, Engert A, Younes A, et al: Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial [published correction appears in J Clin Oncol 2018 Sep 10;36(26):2748]. J Clin Oncol 36(14):1428–1439, 2018. doi:10.1200/JCO.2017.76.0793
5. Chen R, Zinzani PL, Fanale MA, et al: Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J Clin Oncol 35(19):2125–2132, 2017. doi:10.1200/JCO.2016.72.1316
Pronostic du lymphome de Hodgkin
Environ 85 à 90% des patients qui ont un lymphome de Hodgkin classique à stade limité seront guéris, contre 75 à 80% des patients qui ont un stade avancé de la maladie (1). La maladie à stade limité est souvent subdivisée en groupes pronostiques favorables et défavorables. Les maladies à pronostic défavorable sont définies en fonction de facteurs de risque, par exemple
Présence d'une maladie volumineuse
≥ 4 sites ganglionnaire impliqués
Âge > 50 ans
Vitesse de sédimentation (VS) > 50 mm/heure sans symptômes B ou > 30 mm/heure avec symptômes B (perte de poids, fièvre, sueurs nocturnes)
Les facteurs de risque du lymphome de Hodgkin à un stade avancé comprennent
Sexe masculin
Âge > 45 ans
Maladie au stade 4
Signes d'inflammation induits par la tumeur (albumine basse, anémie, leucocytose et lymphopénie).
Cependant, la sélection des facteurs de risque à utiliser pour estimer le pronostic est encore sujette à discussion. La non-obtention d'une rémission complète sous traitement ou une rechute dans les 12 mois est de mauvais pronostic.
Référence pour le pronostic
1. National Institutes of Health: National Cancer Institute Surveillance, Epidemiology, and End-Results (SEER) Program. Cancer Stat Facts—Hodgkin Lymphoma. SEER 2023
Points clés
Le lymphome de Hodgkin est d'origine cellulaire B.
On découvre souvent une lymphadénopathie indolore ou une adénopathie cervicale ou médiastinale sur une rx thorax ou à l'examen clinique.
La biopsie montre des cellules pathognomoniques, binucléées de Reed-Sternberg.
La plupart des patients sont guéris par une chimiothérapie d'association et parfois des thérapies systémiques supplémentaires ou une radiothérapie.
Les options thérapeutiques ultérieures comprennent la chimiothérapie à haute dose, la greffe de cellules souches autologues ou le brentuximab vedotin et les inhibiteurs de point de contrôle (nivolumab et pembrolizumab).
Plus d'information
Ce qui suit est une ressource en anglais qui fournit des informations aux médecins et un soutien et des informations aux patients. LE MANUEL n'est pas responsable du contenu de cette ressource.
Leukemia & Lymphoma Society: Resources for Healthcare Professionals : fournit des ressources de formation pour les professionnels de santé ainsi que des informations pour l'orientation des patients







