







Les bronchectasies sont des dilatations et des destructions des bronches les plus volumineuses dues à une infection et une inflammation chronique. Les causes les plus fréquentes sont la mucoviscidose, les déficits immunitaires et les infections récidivantes. Dans certains cas, elles sont considérées comme idiopathiques. Les symptômes fréquents sont une toux chronique et une expectoration purulente avec ou sans dyspnée. Les symptômes peuvent s'aggraver et peuvent comprendre de la fièvre pendant les exacerbations aiguës. Le diagnostic repose sur l'anamnèse et l'imagerie qui consiste habituellement en une TDM à haute résolution, le diagnostic pouvant cependant être évoqué sur une rx thorax. Le traitement et la prévention des poussées aiguës sont les bronchodilatateurs, l'élimination des sécrétions, les antibiotiques et la prise en charge des complications, telles que l'hémoptysie et d'autres lésions pulmonaires dues à des infections résistantes ou opportunistes. Le traitement des comorbidités est indispensable chaque fois qu'il est possible.
Étiologie des bronchectasies
La bronchectasie doit être considérée comme le point final de divers troubles qui provoquent une inflammation chronique des voies respiratoires fréquent. Les bronchectasies peuvent être
Diffuses: affecte de nombreuses régions des poumons
Focales: n'apparaissant que dans 1 ou 2 régions pulmonaires
Bronchectasie diffuse
Les bronchectasies diffuses se développent le plus souvent en cas d'anomalies génétiques, immunitaires ou anatomiques des voies respiratoires. De nombreux cas apparaissent initialement comme idiopathiques, sans doute en partie parce que l'apparition est si lente que le problème déclenchant peut ne pas être évident au moment où la bronchectasie est reconnue. Cependant, après une enquête utilisant de nouveaux tests génétiques et immunologiques, une étiologie est plus souvent retrouvée dans ces cas idiopathiques.
La mucoviscidose est souvent associée à la bronchectasie diffuse, et la mucoviscidose non diagnostiquée peut représenter jusqu'à 20% des cas idiopathiques. Même les patients hétérozygotes, qui n'ont généralement pas de manifestations cliniques de la mucoviscidose, peuvent avoir un risque accru de bronchectasie.
Les déficits immunitaires comme un déficit immunitaire commun à expression variable peuvent aussi entraîner une atteinte diffuse. L'infection par le virus de l'immunodéficience humaine (VIH) et la dénutrition semblent également augmenter le risque.
De rares anomalies de la structure des voies respiratoires peuvent entraîner une bronchectasie diffuse.
Les anomalies congénitales qui affectent la clairance mucociliaire telles que les syndromes de dyskinésie ciliaire primitive peuvent aussi être en cause, et expliquer ainsi presque 3% de certains cas précédemment considérés idiopathiques.
La bronchectasie diffuse complique parfois des troubles auto-immuns fréquents, comme la polyarthrite rhumatoïde ou le syndrome de Sjögren et peut survenir dans le contexte d'une maladie hématologique maligne, d'une greffe d'organe ou d'un trouble immunologique associé au traitement de ces pathologies. Les bronchectasies peuvent également être liées à des pathologies fréquentes, dont la broncho-pneumopathie chronique obstructive (BPCO), l'asthme, ou les inhalations chroniques, récurrentes.
L'aspergillose bronchopulmonaire allergique est une réaction d'hypersensibilité à Aspergillus spp qui survient le plus souvent chez le sujet asthmatique, mais parfois en cas de mucoviscidose, elle peut être responsable de ou contribuer à des bronchectasies.
Dans les pays dans lesquels la tuberculose est fréquente, la plupart des cas sont dus à la tuberculose, en particulier chez les patients qui ont des fonctions immunitaires altérées en raison d'une malnutrition et d'une infection par le VIH.
Bronchectasie focale
Les bronchectasies localisées sont typiquement secondaires à une pneumonie ou à une obstruction non traitées (p. ex., par des corps étrangers, des tumeurs, des modifications post-chirurgicales, une lymphadénopathie). Des mycobactéries (tuberculeuses ou non tuberculeuses) peuvent à la fois entraîner des bronchectasies focales et coloniser les poumons des patients présentant des bronchectasies d'autre étiologie (voir tableau Facteurs prédisposant aux bronchectasies).
Physiopathologie des bronchectasies
La physiopathologie de la bronchectasie n'est pas entièrement comprise, probablement en partie parce qu'elle représente le point final d'un groupe hétérogène de troubles prédisposant à une inflammation chronique des voies respiratoires. Le modèle le plus largement accepté décrit un "cercle vicieux" d'inflammation, de destruction des voies respiratoires, d'élimination anormale du mucus et d'infection ou de colonisation par des bactéries (1).
Une bronchectasie diffuse apparaît lorsqu'un trouble causal déclenche une inflammation des petites et moyennes voies respiratoires, libérant les médiateurs inflammatoires des neutrophiles intraluminaux. Les médiateurs inflammatoires détruisent l'élastine, le cartilage, et les muscles des grandes voies respiratoires, aboutissant à une bronchodilatation irréversible. Simultanément, dans les voies respiratoires enflammées de petit et moyen calibre, les macrophages et les lymphocytes forment des infiltrats qui épaississent les muqueuses. Cet épaississement provoque l'obstruction des voies respiratoires souvent constatée lors des épreuves fonctionnelles respiratoires.
Avec la progression de la maladie, l'inflammation se propage au-delà des voies respiratoires, ce qui provoque une fibrose du parenchyme pulmonaire environnant. La cause de l'inflammation des petites voies respiratoires dépend de l'étiologie de la bronchectasie. Les contributeurs fréquents sont une altération des mécanismes de libération des voies respiratoires (en raison de la production de mucus épais, visqueux dans la mucoviscidose, d'un trouble de la motilité ciliaire dans la dyskinésie ciliaire primitive, ou de lésions des cils vibratiles et/ou des voies respiratoires secondaires à une infection ou à une lésion) et une diminution des défenses de l'hôte; ces facteurs prédisposent les patients à l'infection et l'inflammation chroniques. Dans le cas d'un déficit immunitaire (en particulier, le déficit immunitaire commun à expression variable) et les causes auto-immunitaires, une inflammation auto-immune peut également avoir un effet contributif.
Une bronchectasie focale se forme habituellement quand les grandes voies respiratoires s'obstruent. L'incapacité à évacuer les sécrétions qui en résulte conduit à un cycle d'infection, inflammation et à des lésions de la paroi des voies respiratoires. Le lobe moyen droit est le plus souvent impliqué car sa bronche est petite et anguleuse et parce que des ganglions lymphatiques se trouvent à proximité. Une adénopathie due à une infection mycobactérienne provoque parfois une obstruction bronchique et une bronchectasie focale.
L'inflammation continue modifiant l'anatomie des voies respiratoires, des bactéries pathogènes (y compris parfois des mycobactéries), colonisent les voies respiratoires. Les microrganismes fréquents comprennent
Haemophilus influenzae
Moraxella catarrhalis
Pseudomonas aeruginosa
Staphylococcus aureus
Streptococcus pneumoniae
Mycobactéries non tuberculeuses
La colonisation par S. aureus est fortement associée à la mucoviscidose; une culture positive à S. aureus doit faire évoquer une mucoviscidose non diagnostiquée. En outre, la colonisation par P. aeruginosa tend à indiquer une maladie grave et prédit des résultats plus défavorables, dont un risque accru d'exacerbations, d'hospitalisation, de mauvaise qualité de vie, de déclin rapide de la fonction pulmonaire et de décès. La colonisation par de multiples microrganismes est fréquente, et la résistance aux antibiotiques représente une préoccupation chez les patients chez qui sont nécessaires des cycles fréquents d'antibiotiques lors des exacerbations.
Complications
Lorsque la maladie progresse, l'inflammation et l'hypoxémie chroniques induisent une néovascularisation des artères bronchiques (et non pas les artères pulmonaires). Les parois de l'artère bronchique se rompent facilement, ce qui provoque une hémoptysie, qui peut être massive et menacer le pronostic vital. D'autres complications vasculaires comprennent l'hypertension pulmonaire due à une vasoconstriction, l'artérite, et parfois de shunt bronchique à partir des vaisseaux pulmonaires.
La colonisation par les microrganismes multirésistants peut induire une inflammation chronique de bas grade des voies respiratoires. Cette inflammation peut évoluer provoquant des exacerbations récurrentes et aggraver les limitations du flux aérien au cours des épreuves fonctionnelles respiratoires.
Référence pour la physiopathologie
1. Cole PJ: Inflammation: a two-edged sword—the model of bronchiectasis. Eur J Respir Dis Suppl 147:6–15, 1986.
Symptomatologie de la bronchectasie
Les symptômes débutent généralement de façon insidieuse et s'aggravent progressivement au fil des ans, accompagnés d'épisodes d'aggravation aiguë.
Le symptôme le plus fréquent est une toux chronique qui produit des crachats épais, abondants et souvent purulents. Une dyspnée et un wheezing sont fréquents, et une douleur thoracique de type pleural peut se développer. Dans les cas avancés, l'hypoxémie et l'insuffisance cardiaque droite dues à l'hypertension pulmonaire peuvent augmenter la dyspnée. L'hémoptysie, qui peut être massive, est due à une néovascularisation des voies respiratoires.
Les exacerbations aiguës sont fréquentes et entraînent souvent d'une infection nouvelle ou qui s'aggrave. Les poussées aiguës de la maladie sont marquées par une aggravation de la toux, une augmentation de la dyspnée ainsi que du volume et de la purulence des expectorations. Une fièvre peu élevée et des symptômes constitutionnels (p. ex., fatigue, sensation de malaise) peuvent également être présents.
L'halitose et des anomalies auscultatoires pulmonaires comprenant des râles crépitants, des ronchi et un wheezing, sont des signes d'examen clinique caractéristiques. Un hippocratisme digital est rare mais peut être présent. Une rhinosinusite et des polypes nasaux chroniques peuvent être présents, en particulier en cas de mucoviscidose ou de dyskinésie ciliaire primitive. La masse maigre diminue souvent, peut-être en raison de l'inflammation et des excès de cytokines et, en cas de mucoviscidose, en raison de la malabsorption.
Diagnostic de la bronchectasie
Rx thorax
TDM du thorax à haute résolution
Épreuves fonctionnelles respiratoires afin d'évaluer la fonction respiratoire de base et suivre l'évolution de la maladie
Culture des expectorations à la recherche de bactéries et de mycobactéries pour identifier les microrganismes colonisateurs
Examens spécifiques pour le bilan étiologique
Le diagnostic repose sur l'anamnèse, l'examen clinique et rx, avec initialement une rx thorax. La bronchite chronique peut présenter des similitudes cliniques avec la bronchectasie, mais cette dernière s'en distingue par une augmentation de la purulence et du volume des crachats quotidiens et un aspect des bronches dilaté à l'imagerie.
Imagerie
La rx thorax est généralement anormal et peut être diagnostique. Les signes radiographiques évocateurs d'une bronchectasie sont un épaississement des parois des voies respiratoires et/ou une dilatation des voies respiratoires; les signes typiques comprennent des densités périhilaires linéaires floues avec indistinction des artères pulmonaires centrales, des anneaux flous dus à des voies respiratoires épaissies vues en coupe (parallèles au faisceau de rx), et des aspects en "rails de tram" causés par des voies respiratoires épaissies dilatées perpendiculaires au faisceau rx. Des voies respiratoires dilatées remplies de bouchons muqueux peuvent aussi causer des opacités dispersées, tubulaires allongées.
Les aspects rx peuvent différer selon la maladie sous-jacente: les bronchectasies de la mucoviscidose se développent préférentiellement dans les lobes supérieurs, alors que les bronchectasies dues à une obstruction endobronchique provoquent des anomalies rx plus focales.
La TDM à haute résolution est l'examen de choix pour évaluer l'étendue des bronchectasies et présente une grande sensibilité et spécificité. Les signes TDM typiques comprennent une dilatation des voies respiratoires (dans lesquelles la lumière interne de 2 ou plusieurs voies respiratoires dépasse le diamètre de l'artère adjacente) et le signe de la chevalière dans lequel des voies respiratoires dilatées dont la paroi est épaissie apparaissant adjacente à une artère de plus petit diamètre en incidence transaxiale. L'absence de rétrécissement normal des bronches peut mettre en évidence des bronches de taille moyenne qui s'étendent pratiquement jusqu'à la plèvre. Les "lignes de tram" sont facilement visibles sur la TDM.
Les lésions des voies respiratoires augmentant avec le temps, les bronchectasies de cylindriques deviennent variqueuses avec ensuite des signes kystiques sur l'imagerie. Atélectasies, condensations, bouchons muqueux et diminution de la vascularisation sont des aspects non spécifiques. Dans les bronchectasies par traction, la fibrose pulmonaire tire ou déforme les voies respiratoires de telle manière que cela simule la bronchectasie sur l'imagerie.

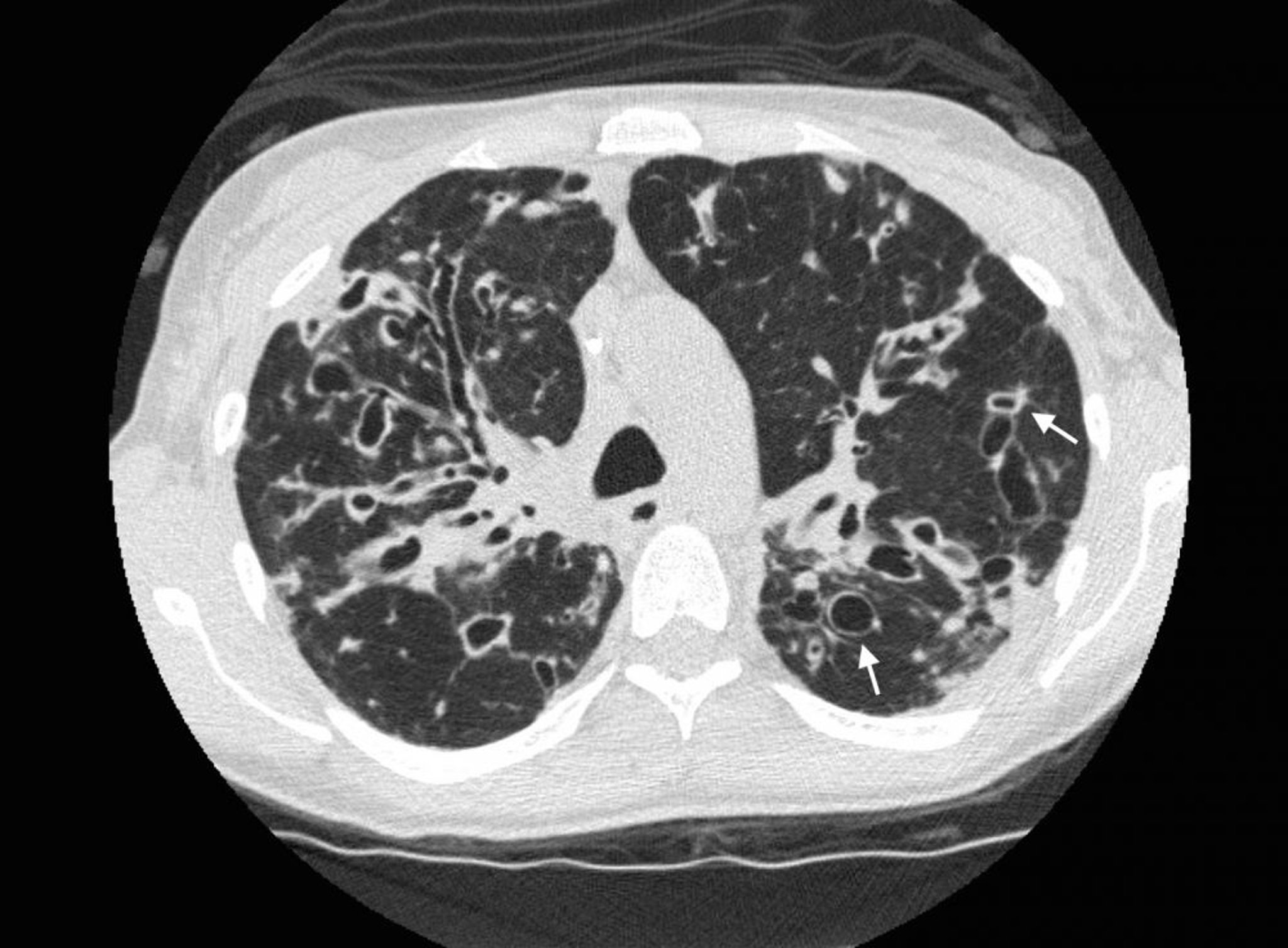
Photo courtoisie de Başak Çoruh, MD.

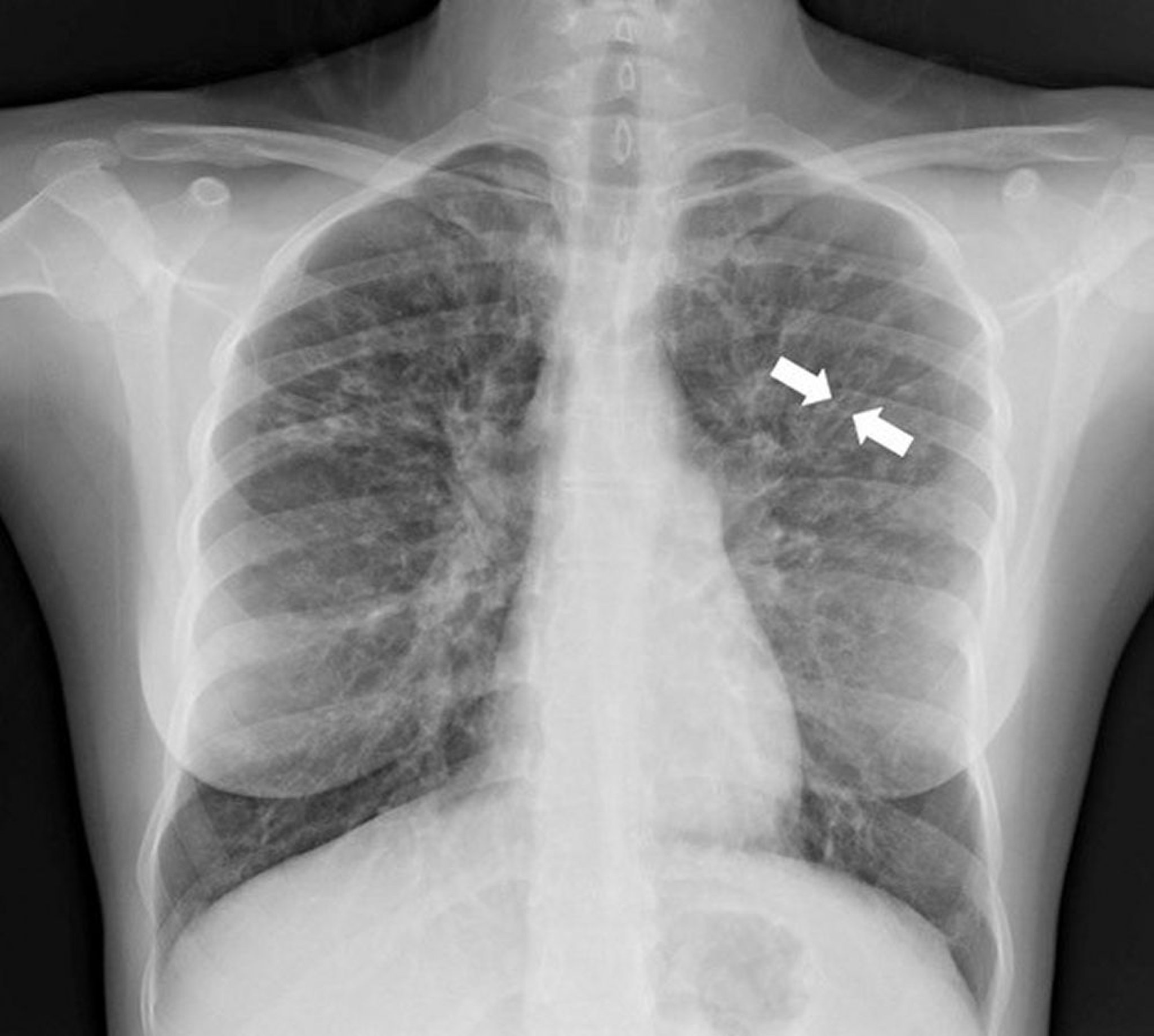
Photo courtoisie de Başak Çoruh, MD.
Épreuves fonctionnelles respiratoires
Les épreuves fonctionnelles respiratoires permettent d'évaluer la fonction respiratoire de base et de suivre l'évolution de la maladie. Les bronchectasies entraînent une obstruction bronchique (diminution du rapport volume expiratoire maximal en 1 s [VEMS1] sur la capacité vitale forcée [CVF], et du VEMS1/CVF); le VEMS1 peut être amélioré en réponse aux bronchodilatateurs bêta-agonistes. Dans les cas plus avancés, la fibrose progressive peut entraîner une diminution de la capacité vitale forcée (CVF), une anomalie restrictive aux mesures du volume pulmonaire et une diminution de la capacité de diffusion du monoxyde de carbone (DLCO).
Diagnostic étiologique
Pendant une période sans exacerbation, il faut mettre en culture des expectorations spontanées ou induites pour déterminer quelles sont les bactéries prédominantes et leurs sensibilités. Cette information aide à choisir les antibiotiques au cours des exacerbations.
Une numération formule sanguine peut permettre de déterminer la gravité de l'activité de la maladie et d'identifier une éosinophilie, ce qui peut suggérer le diagnostic de complications.
Les colorations et cultures d'expectoration pour rechercher des microrganismes bactériens, mycobactériens (Mycobacterium avium complex et M. tuberculosis), et fongiques (Aspergillus spp) peuvent également permettre d'identifier les causes de l'inflammation chronique des voies respiratoires.
Une infection mycobactérienne non tuberculeuse cliniquement significative est diagnostiquée par la mise en évidence d'un nombre élevé de colonies de ces mycobactéries dans les cultures de crachats en série ou de liquide de lavage broncho-alvéolaire chez les patients qui ont des granulomes à la biopsie ou des signes radiologiques de maladie concomitante.
Lorsque la cause de la bronchectasie est incertaine, des examens supplémentaires fonctions de l'anamnèse et de l'imagerie peuvent être effectués. Les tests peuvent comprendre:
Taux d'alpha-1-antitrypsine à évaluer pour rechercher un déficit en alpha-1-antitrypsine si la TDM à haute résolution montre un emphysème du lobe inférieur
Le facteur rhumatoïde, les AAN (anticorps antinucléaires), et les anticorps anti-cytoplasme de polynucléaires neutrophiles si une maladie auto-immune est évoquée
Immunoglobulines sériques (IgG IgA, IgM) et électrophorèse sérique pour diagnostiquer un déficit immunitaire commun à expression variable
Taux de précipitines d'Aspergillus et d'IgE sériques en cas d'éosinophilie, pour éliminer l'aspergillose bronchopulmonaire allergique
Des tests de la sueur (un test positif doit être confirmé par la répétition du test) et une analyse de la mutation du gène CFTR pour diagnostiquer la mucoviscidose (y compris chez l'adulte de > 40 ans sans cause identifiable de dilatation des bronches, surtout en cas d'implication du lobe supérieur, de malabsorption, ou d'infertilité masculine)
Les patients qui ont des preuves biologiques d'immunodéficience doivent être adressés à un spécialiste en immunologie pour évaluation parce que les résultats des tests sont souvent difficiles à interpréter. Des examens complémentaires supplémentaires peuvent également être nécessaires pour confirmer le type de déficit immunitaire présent et déterminer les options de traitement.
La dyskinésie ciliaire primitive ne doit être évoquée que si les adultes qui ont une bronchectasie ont aussi une maladie chronique des sinus ou une otite moyenne, en particulier si les problèmes ont persisté depuis l'enfance. La bronchectasie chez ces patients peut être prédominante au niveau du lobe moyen droit et au niveau lingulaire, et une infertilité masculine ou une dextrocardie peuvent être présentes. Le taux nasal ou oral d'oxyde nitrique expiré est souvent bas. Le diagnostic de certitude nécessite l'examen d'un prélèvement nasal ou bronchique pour mettre en évidence la structure anormale de l'épithélium ciliaire en microscopie électronique à transmission.
Le diagnostic de dyskinésie ciliaire primitive doit normalement être posé dans des centres spécialisés car le bilan peut être difficile à établir. Des anomalies structurelles non spécifiques peuvent être présentes dans jusqu'à 10% des cils chez les sujets en bonne santé et chez les patients qui ont une maladie pulmonaire; une infection peut provoquer une dyskinésie transitoire. L'ultrastructure ciliaire peut aussi être normale chez certains patients qui ont des syndromes de dyskinésie ciliaire primitive, nécessitant d'autres examens pour identifier la fonction ciliaire anormale.
La bronchoscopie est indiquée en cas de suspicion de lésion obstructive ou anatomique.
Définition et évaluation des exacerbations
L'exacerbation d'une bronchectasie est définie comme un patient qui présente une bronchectasie avec détérioration pendant au moins 48 heures dans ≥ 3 des symptômes suivants (1):
Essoufflement et/ou intolérance à l'effort
Toux
Fatigue et/ou sensation de malaise
Hémoptysie
Purulence des expectorations
Volume et/ou consistance des expectorations
Le degré du dépistage dépend de la gravité du tableau clinique. En cas d'exacerbations légères à modérées, répéter les cultures d'expectorations pour confirmer les microrganismes en cause et leur sensibilité peut être suffisant. Ces examens permettent une couverture antibiotique plus précise et d'exclure les pathogènes opportunistes.
Chez les patients les plus gravement atteints, un NFS, une rx thorax, et éventuellement d'autres examens il peut être justifié d'exclure des complications fréquentes de l'infection pulmonaire grave, comme un abcès du poumon et un empyème.
Référence pour le diagnostic
1. Hill AT, Haworth CS, Aliberti S, et al: Pulmonary exacerbation in adults with bronchiectasis: A consensus definition for clinical research. Eur Respir J 49:1700051, 2017.
Traitement de la bronchectasie
Prévention des exacerbations par des vaccinations régulières et parfois par des antibiotiques suppresseurs
Mesures pour éliminer les sécrétions respiratoires
Bronchodilatateurs et parfois corticostéroïdes inhalés en cas d'obstruction bronchique réversible
Antibiotiques et bronchodilatateurs lors des poussées aiguës
Traitement précoce par des antiviraux de toute infection virale, en particulier de la grippe et du COVID-19
Parfois résection chirurgicale en cas de maladie localisée avec symptômes ou saignement réfractaires
Transplantation pulmonaire chez des patients soigneusement sélectionnés qui ont une maladie avancée malgré un traitement maximal
Les objectifs du traitement clés visent à contrôler les symptômes et à améliorer la qualité de vie, réduire la fréquence des poussées et préserver la fonction pulmonaire (1, 2).
Comme pour les patients qui ont une maladie pulmonaire chronique, les recommandations comprennent les suivantes:
Vaccination contre la grippe annuelle
Les techniques de dégagement des voies respiratoires sont utilisées pour réduire la toux chronique en cas de production d'expectorations significative et d'obstruction par du mucus et réduire les symptômes au cours des exacerbations. Ces techniques comprennent l'exercice régulier, la kinésithérapie thoracique avec drainage postural et la percussion thoracique, les dispositifs de pression expiratoire positive, les ventilateurs à percussions intrapulmonaires, les vestes pneumatiques et le drainage autogène (une technique de respiration considérée comme aidant à mobiliser les sécrétions des voies respiratoires périphériques vers les voies respiratoires centrales). Les patients doivent être formés à ces techniques par un kinésithérapeute spécialiste des problèmes respiratoires et doivent utiliser la technique la plus efficace pour eux et celle à laquelle ils adhèrent le mieux compte-tenu de l'absence de preuve permettant de privilégier telle ou telle technique. Les patients doivent être informés qu'ils doivent continuer à pratiquer leurs techniques de dégagement des voies respiratoires pendant au moins 10 minutes; ils peuvent s'arrêter quand ils produisent 2 toux claires (3) ou après 30 minutes.
Dans le cas des patients présentant une obstruction réversible des voies respiratoires, le traitement bronchodilatateur (p. ex., par une association bêta-adrénergique à longue durée d'action, un antagoniste muscarinique à longue durée d'action et un médicament bêta-adrénergique à action rapide comme indiqué par les symptômes et la gravité de l'obstruction, tel qu'il est utilisé en cas de BPCO) peut permettre d'améliorer la fonction pulmonaire et la qualité de la vie. Les corticostéroïdes inhalés peuvent également être utilisés en cas d'exacerbations fréquentes ou de variabilité importante des mesures de la fonction pulmonaire (c'est-à-dire, obstruction réversible des voies respiratoires après l'administration d'un bronchodilatateur), mais leur rôle est controversé. La rééducation fonctionnelle respiratoire peut être utile. Maintenir une hydratation adéquate est également important.
Les patients mucoviscidosiques peuvent recevoir une variété de traitements par nébulisation, dont un mucolytique (rhDNase, également appelée dornase alpha) et un sérum hypertonique (7%), peuvent réduire la viscosité des crachats et améliorer le dégagement des voies respiratoires. Chez les patients qui n'ont pas de mucoviscidose, il n'y a pas de preuve évidente de leur bénéfice, de sorte que l'humidification et la solution physiologique sont recommandées comme traitements inhalatoires. La rhDNase inhalée peut être nocive en cas de bronchectasie non provoquée par la mucoviscidose.
Il n'y a pas de consensus sur la meilleure stratégie antibiotique pour éviter ou limiter la fréquence des poussées aiguës. L'utilisation régulière d'antibiotiques suppresseurs ou selon un calendrier de rotation réduit les symptômes et les exacerbations, mais peut augmenter le risque que les infections futures impliquent des microrganismes résistants. Les directives actuelles suggèrent l'utilisation d'antibiotiques en cas de ≥ 3 exacerbations par an et peut-être aussi en cas de nombre d'exacerbations moindre en présence d'une colonisation par P. aeruginosa. Certaines lignes directrices suggèrent une tentative d'éradication de microrganismes tels que P. aeruginosa ou S. aureus quand ils sont détectés pour la première fois dans des cultures d'expectorations (3).
Le traitement chronique par les macrolides réduit les exacerbations aiguës en cas de bronchectasie et peut ralentir le déclin de la fonction pulmonaire en cas de mucoviscidose (4, 5, 6). Les macrolides sont considérés comme bénéfiques principalement en raison de leurs effets anti-inflammatoires ou immunomodulateurs.
Les antibiotiques inhalés (p. ex., amikacine, aztréonam, ciprofloxacine, gentamicine, tobramycine) peuvent réduire la charge bactérienne des expectorations et réduire la fréquence des exacerbations. Les preuves à l'appui de leur utilisation et de leurs avantages sont les plus solides dans la population des patients mucoviscidosiques.
Les patients qui ont une bronchectasie et qui développent une grippe ou une infection par le SARS-CoV-2 doivent être traités par des médicaments antiviraux pour prévenir les complications et le développement d'une forme grave. Dans le cas de la grippe, le traitement repose généralement sur l'oseltamivir quelle que soit la durée des symptômes viraux. Les infections par le SARS-CoV-2 doivent être traitées dès que possible en cas de bronchectasie, en particulier en cas d'immunosuppression sous-jacente. Le protocole préféré est le nirmatrelvir augmenté par le ritonavir, débuté dans les 5 jours suivant l'apparition des symptômes, mais d'autres options sont disponibles. Les corticostéroïdes oraux ne sont pas recommandés chez les patients ambulatoires porteurs de COVID-19 en l'absence d'une maladie concomitante, telle que l'asthme ou la BPCO, qui est connue pour bénéficier des corticostéroïdes. Les patients qui ont une bronchectasie doivent être étroitement surveillés à la recherche d'une co-infection bactérienne et toute exacerbation déclenchée par une infection virale doit être traitée comme décrit ci-dessus.
Les séances de traitement de la bronchectasie doivent être effectuées dans l'ordre suivant:
Bronchodilatateurs inhalés à courte durée d'action
Thérapie mucolytique (si prescrite)
Technique de libération des voies respiratoires
Tous les antibiotiques prescrits inhalés ou nébulisés, les bronchodilatateurs à longue durée d'action ou les corticostéroïdes
Les pathologies sous-jacentes doivent être traitées pour ralentir la progression de la maladie pulmonaire.
En cas de déficit immunitaire sous-jacent: immunoglobulines intraveineuses programmées (qui peuvent réduire la fréquence des infections des voies respiratoires inférieures [7])
Chez les patients qui ont une mucoviscidose: des antibiotiques et des bronchodilatateurs inhalés ainsi qu'un soutien complet et une supplémentation diététique. La plupart des patients atteints de mucoviscidose tirent profit d'un traitement modulateur de CFTR qui peut diminuer les exacerbations. Les patients qui ont une mucoviscidose doivent recevoir tout ou partie de leurs soins par des équipes spécialisées dans la mucoviscidose, généralement dans un centre de soins spécialisé.
En cas d'aspergillose bronchopulmonaire allergique: corticostéroïdes et parfois antifongiques azolés
En cas de déficit en alpha-1-antitrypsine: traitement de substitution
Aggravations aiguës
Les exacerbations aiguës sont traitées par des antibiotiques, des bronchodilatateurs inhalés (particulièrement en cas de wheezing), et par des tentatives augmentées d'élimination du mucus, en utilisant des techniques mécaniques, traitement de la déshydratation (le cas échéant), l'humidification et la nébulisation d'une solution physiologique (et des mucolytiques en cas de mucoviscidose). Les corticostéroïdes inhalés ou oraux sont fréquemment administrés pour traiter l'inflammation et l'aggravation de l'obstruction des voies respiratoires. Le choix des antibiotiques dépend des résultats des cultures précédentes et de l'existence ou non d'une mucoviscidose (8).
Les antibiotiques initiaux dans la mucoviscidose en l'absence de résultats de culture doivent être efficaces contre H. influenzae, M. catarrhalis, S. aureus, et S. pneumoniae. Les exemples comprennent l'amoxicilline/acide clavulanique, l'azithromycine, la clarithromycine, et le triméthoprime/sulfaméthoxazole. Les patients qui ont une colonisation connue par P. aeruginosa ou de plus graves exacerbations doivent recevoir des antibiotiques efficaces contre ce microrganisme (p. ex., de la ciprofloxacine ou de la lévofloxacine) jusqu'à ce que les résultats des cultures répétées soient disponibles. Les antibiotiques doivent être ajustés en fonction des résultats de la culture et sont administrés pendant une durée habituelle de 14 jours, en particulier si P. aeruginosa est détecté. Des cycles plus courts sont réservés aux patients atteints d'une maladie modérée.
Le choix initial des antibiotiques dans la mucoviscidose est guidé par le résultat des cultures d'expectorations précédentes (effectuées régulièrement chez tous les patients atteints de mucoviscidose). Chez les enfants, les microrganismes les plus fréquents sont S. aureus et H. influenzae et les quinolones telles que la ciprofloxacine et la lévofloxacine peuvent être utilisées. Dans les mucoviscidoses évoluées, les infections impliquent des souches hautement résistantes de certains microrganismes Gram négatifs comme P. aeruginosa, Burkholderia cepacia, et Stenotrophomonas maltophilia. En cas d'infection provoquée par ces microrganismes, le traitement repose sur plusieurs antibiotiques (p. ex., tobramycine, aztréonam, ceftazidime, céfépime). Une administration IV est nécessaire.
Complications
Les hémoptysies importantes sont généralement traitées par embolisation des artères bronchiques, mais la résection chirurgicale peut être envisagée si l'embolisation n'est pas efficace et si la fonction respiratoire le permet.
Les surinfections par des mycobactéries atypiques telles que le complexe M. avium nécessitent presque toujours des protocoles antibiotiques multiples qui comprennent la clarithromycine ou l'azithromycine, la rifampicine ou la rifabutine et l'éthambutol. Le traitement antibiotique est généralement poursuivi jusqu'à ce que les cultures des crachats soient négatives pendant 12 mois.
La résection chirurgicale est rarement indiquée, mais elle peut être envisagée lorsque la bronchectasie est localisée malgré un traitement médical optimisé et lorsque les symptômes sont intolérables. Chez certains patients qui présentent des bronchectasies diffuses en particulier en cas de mucoviscidose, la transplantation pulmonaire est une option.
Références pour le traitement
1. Polverino E, Gemine PC, McDonnell MJ, et al: European Respiratory Society guidelines for the management of adult bronchiectasis. Eur Respir J 50: 1700629, 2017. doi: 10.1183/13993003.00629-2017
2. Nicholson CH, Holland AE, Lee AL: The Bronchiectasis Toolbox - A Comprehensive Website for the Management of People with Bronchiectasis. Med Sci (Basel) 5, 13, 2017.
3. Hill AT, Sullivan AL, Chalmers JD, et al: British Thoracic Society Guideline for bronchiectasis in adults. Thorax 74(Suppl 1):1–69, 2019. doi: 10.1136/thoraxjnl-2018-212463
4. Wong C, Jayaram L, Kraals N, et al: Azithromycin for the prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): A randomised, double blind, placebo controlled trial. Lancet 380: 660–667, 2012.
5. Altenburg J, de Graaf CS, Stienetra Y, et al: Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: The BAT randomized controlled trial. JAMA 309: 1251–1259, 2013.
6. Serisier DJ, Martin ML, McGuckin MA, et al: Effect of long-term, low dose erythromycin on pulmonary exacerbations among patients with non-cystic fibrosis bronchiectasis: the BLESS randomized controlled trial. JAMA 309: 1260–1267, 2013.
7. Quinti I, Sorellina A, Guerra A, et al: Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: Results from a multicenter prospective cohort trial. J Clin Immunol 31: 315–322, 2011.
8. Flume PA, Mogayzel PJ Jr, Robinson KA, et al: Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. Am J Respir Crit Care Med 80:802–808, 2009. doi: 10.1164/rccm.200812-1845PP
Pronostic de la bronchectasie
Le pronostic varie largement. La baisse annuelle moyenne du VEMS1 est d'environ 50 à 55 mL (la diminution normale chez le sujet en bonne santé est d'environ 20 à 30 mL). Les patients qui ont une mucoviscidose ont eu historiquement le pronostic le plus défavorable, avec une médiane de survie de 36 ans. Cependant, l'avènement des modulateurs de CFTR (cystic fibrosis transmembrane regulator) a entraîné des améliorations significatives de l'évolution, même en cas de maladie pulmonaire avancée (1).
Référence pour le pronostic
1. Shteinberg M, Taylor-Cousar JL: Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur Respir Rev 29(155):190112, 2020. doi: 10.1183/16000617.0112-2019
Points clés
Dans les bronchectasies, l'inflammation chronique de diverses causes détruit l'élastine, le cartilage et le muscle des grandes voies respiratoires, entraînant des lésions irréversibles et des voies respiratoires dilatées chroniquement colonisées par des microrganismes infectieux.
Les patients ont une toux chronique productive avec exacerbations aiguës intermittentes.
Le diagnostic est établi par l'imagerie, habituellement la TDM; des cultures doivent être effectuées pour identifier le(s) microrganisme(s) colonisateur(s).
Prévenir les exacerbations en utilisant les vaccinations appropriées, des mesures de dégagement des voies respiratoires, et parfois des antibiotiques macrolides.
Traiter les exacerbations par des antibiotiques, des bronchodilatateurs, des mesures plus fréquentes de dégagement des voies respiratoires et parfois des corticostéroïdes.







