







La mucoviscidose est une maladie héréditaire des glandes exocrines, touchant essentiellement le tube digestif et l'appareil respiratoire. Elle entraîne une pneumopathie chronique, une insuffisance pancréatique exocrine, une maladie hépatobiliaire et une concentration excessive d'électrolytes dans la sueur. Le diagnostic repose sur le test de la sueur ou l'identification de 2 variantes géniques responsables de la mucoviscidose chez des patients présentant un résultat de test de dépistage néonatal positif ou des signes cliniques caractéristiques. Le traitement est un traitement de support qui repose sur des soins multidisciplinaires agressifs avec des petites molécules correctrices et des agents de potentialisation qui ciblent l'anomalie de la protéine CFTR (cystic fibrosis transmembrane conductance regulator).
La mucoviscidose est une maladie génétique potentiellement mortelle qui, aux États-Unis, est observée chez environ 1/3300 naissances blanches, 1/15 300 naissances noires et 1/32 000 naissances asiatiques-américaines. Environ 40 000 personnes sont atteintes de mucoviscidose aux États-Unis et environ 100 000 personnes sont atteintes de mucoviscidose dans le monde. Grâce à l'amélioration du traitement et de l'espérance de vie, environ 58% des patients atteints de muscovicidose aux États-Unis sont à présent des adultes (1).
Références générales
1. Cystic Fibrosis Foundation Patient Registry 2021 Annual Data Report Bethesda, Maryland 2022 Cystic Fibrosis Foundation. Consulté le 20 octobre 2023.
Étiologie de la mucoviscidose
La mucoviscidose est une maladie génétique autosomique récessive touchant, à l'état hétérozygote, 3% environ des populations blanches. Le gène responsable a été localisé sur le bras long du chromosome 7. Il code une protéine membranaire appelée Cystic Fibrosis Transmembrane Regulator (CFTR). Le variant génique le plus fréquent, F508del, est observé dans environ 85% des allèles mutés de la mucoviscidose; > 2000 variants plus rares du gène CFTR ont été identifiées.
Le CFTR est un canal Cl régulé par l'AMPc (cyclic adenosine monophosphate), qui permet le transport des ions Cl, sodium et bicarbonate à travers les membranes épithéliales. Il possède probablement un certain nombre d'autres fonctions. La maladie ne se manifeste que chez les homozygotes. Les hétérozygotes sont cliniquement indemnes, mais peuvent présenter de discrètes anomalies du transport ionique transmembranaire.
Les variants du CFTR ont été répartis en 6 catégories, en fonction de la façon dont le variant affecte la fonction ou le métabolisme de la protéine CFTR. Les patients présentant des variants de classe I, II ou III sont considérés comme ayant un génotype plus sévère, qui se traduit par une fonction du gène CFTR faible ou inexistante, alors que les patients présentant 1 ou 2 variants de classe IV, V ou VI sont considérés avoir un génotype moins sévère qui se traduit par une fonction résiduelle du CFTR. Cependant, il n'y a pas de relation stricte entre des variants spécifiques et les manifestations de la maladie, les tests cliniques (c'est-à-dire, de la fonction des organes) plutôt que le génotypage est un meilleur guide pronostique. Les variants de CFTR peuvent être correspondre à des mutations frameshift (une délétion ou une insertion dans une séquence d'ADN qui modifie la façon dont une séquence est lue) ou des mutations non-sens (stop).
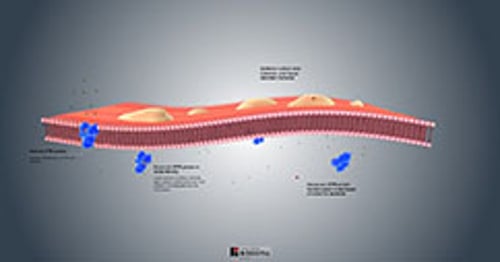
Physiopathologie de la mucoviscidose
Pratiquement toutes les glandes exocrines sont atteintes avec une répartition et des degrés de gravité variables. Les glandes peuvent
Etre obstruées par du mucus visqueux présent dans la lumière (pancréas, glandes intestinales, voies biliaires intrahépatiques, vésicule biliaire et glandes sous-maxillaires)
Apparaître histologiquement anormales et produire une quantité excessive de sécrétions (glandes trachéo-bronchique et de Brunner)
Apparaître histologiquement normales mais sécréter des quantités excessives de sodium et de Cl (glandes sudorales, parotides, et petites glandes salivaires)
Respiratoire
Bien que les poumons soient généralement histologiquement normaux à la naissance, la plupart des patients développent des signes d'une maladie pulmonaire débutant dans l'enfance ou à la petite enfance. La constitution de bouchons muqueux (dans les bronches) et l'infection bactérienne chronique, accompagnées d'une réponse inflammatoire exagérée, lèsent les voies respiratoires, aboutissant à des bronchectasies et à une insuffisance respiratoire. L'évolution est caractérisée par des exacerbations, associant infection et inflammation, induisant une dégradation progressive de la fonction respiratoire.
Les lésions pulmonaires sont probablement la conséquence de l'obstruction diffuse des bronchioles par des sécrétions muqueuses anormalement épaisses. La bronchiolite et l'obstruction par des sécrétions mucopurulentes des voies respiratoires sont secondaires à une obstruction et à une infection. L'inflammation chronique provoquée par la libération de protéases et de cytokines proinflammatoires par les cellules des voies respiratoires contribue aussi aux lésions pulmonaires. L'altération des voies respiratoires est plus fréquente que les modifications du parenchyme pulmonaire et l'emphysème n'est pas le plus fréquent. Environ 50% des patients ont une hyperréactivité bronchique qui peut être sensible aux bronchodilatateurs.
Chez les patients qui ont une maladie pulmonaire avancée, l'hypoxémie chronique entraîne une hypertrophie de la paroi musculaire des artères pulmonaires entraînant une hypertension artérielle pulmonaire et une hypertrophie ventriculaire droite.
Les poumons de la plupart des patients sont colonisés par des bactéries pathogènes. Au début de l'évolution, Staphylococcus aureus est l'agent pathogène le plus fréquent, mais avec l'évolution de la maladie, Pseudomonas aeruginosa est le plus souvent isolé. Une variante mucoïde de P. aeruginosa est très spécifiquement associée à la mucoviscidose et a un plus mauvais pronostic que P. aeruginosa non mucoïde.
Aux États-Unis, la prévalence de S. aureus résistant à la méthicilline (SARM) dans les voies respiratoires est à présent d'environ 25%; les patients chroniquement infectés par le SARM ont un déclin plus rapide de la fonction pulmonaire et des survies plus faibles que les patients qui ne le sont pas.
La colonisation par le complexe Burkholderia cepacia concerne jusqu'à 2-3% des patients adultes et peut s'accompagner d'une détérioration pulmonaire rapide.
Les mycobactéries non tuberculeuses, dont le complexe Mycobacterium avium et M. abscessus, sont des agents pathogènes respiratoires potentiels. La prévalence est d'environ 14% et varie avec l'âge et la situation géographique. La distinction entre colonisation et infection peut être difficile à établir.
D'autres agents pathogènes respiratoires fréquents sont Stenotrophomonas maltophilia, Achromobacter xylosoxidans et Aspergillus spp.
Des bactéries anaérobies et des virus respiratoires communs sont fréquemment présents dans les voies respiratoires des patients atteints de mucoviscidose, mais leur rôle dans la progression de la maladie n'a pas été bien établi.
Gastrointestinal
Le pancréas, l'intestin et le système hépatobiliaire sont fréquemment affectés dans cette maladie. La fonction pancréatique exocrine est altérée chez 85 à 95% des patients. Une exception est un sous-ensemble de patients qui ont certaines variantes de CFTR avec fonction résiduelle, chez lesquels la fonction pancréatique est préservée. Les patients insuffisants pancréatiques ont une malabsorption des graisses, des vitamines liposolubles et des protéines. Le contenu duodénal est anormalement visqueux avec une diminution ou une absence de l'activité enzymatique et de la concentration en bicarbonate; les taux de trypsine et de chymotrypsine dans les selles sont nuls ou abaissés. La dysfonction endocrine du pancréas est moins fréquente, mais une diminution de la tolérance au glucose ou un diabète sucré sont présents chez environ 2% des enfants, 20% des adolescents, et jusqu'à 50% des adultes.
L'atteinte des voies biliaires avec une stase biliaire et une obstruction des voies biliaires induit une fibrose hépatique asymptomatique chez 30% des patients. Environ 3 à 4% des patients progressent vers une cirrhose biliaire avec hypertension portale et varices, habituellement vers l'âge de 12 ans. L'insuffisance hépatocellulaire est un événement rare et tardif. Il existe une incidence accrue de calculs biliaires, qui est habituellement asymptomatique.
Des sécrétions intestinales anormalement visqueuses peuvent être la cause d'iléus méconial chez le nouveau-né et parfois d'occlusions coliques par bouchon méconial. Les enfants et les adultes plus âgés peuvent également présenter une constipation chronique ou une occlusion intestinale.
D'autres problèmes gastro-intestinaux et hépatiques peuvent survenir tels qu'une invagination intestinale aiguë, un volvulus, un prolapsus rectal, un abcès appendiculaire, une pancréatite, un risque accru de cancer des voies hépatobiliaires et de cancers gastro-intestinaux (dont le pancréas), de reflux gastro-œsophagien et une augmentation de la prévalence de la maladie de Crohn et de maladie cœliaque.
Autres
Une infertilité est observée chez 98% des hommes adultes qui présentent un défaut de développement des canaux déférents ou d'autres formes d'azoospermie obstructive. Chez la femme, la fertilité est un peu diminuée par une viscosité excessive des sécrétions cervicales, bien que nombre de femmes puissent mener des grossesses à terme. L'évolution de la grossesse pour la mère et le nouveau-né est liée à la santé de la mère.
D'autres complications comprennent la rhinosinusite chronique, l'ostéopénie/ostéoporose, la dépression et l'anxiété, des douleurs chroniques, une apnée obstructive du sommeil, d'autres troubles du sommeil, des calculs rénaux, une maladie rénale chronique dépendant de la dialyse (éventuellement liée aux traitements ainsi qu'à la mucoviscidose), une anémie ferriprive, une surdité neurosensorielle et acouphènes dus à une exposition à des médicaments ototoxiques (en particulier des aminosides) et des arthralgies/arthrites épisodiques.
Symptomatologie de la mucoviscidose
Respiratoire
Cinquante pour cent des patients non diagnostiqués à la naissance, se présentent initialement avec des manifestations pulmonaires, débutant souvent au cours de la petite enfance. Des infections bronchiques chroniques ou récidivantes, manifestées par de la toux, une expectoration et par un wheezing, sont fréquentes. La toux est le symptôme chronique le plus fréquent, souvent accompagnée de crachats, de vomissements et de troubles du sommeil. Des rétractions intercostales, une mise en jeu des muscles respiratoires accessoires, une déformation thoracique en tonneau, un hippocratisme digital, une cyanose et une diminution de la tolérance à l'effort avec l'évolution de la maladie. L'atteinte des voies respiratoires hautes comprend une polypose nasale et une rhinosinusite chronique ou récidivante.
Les complications pulmonaires comprennent le pneumothorax, l'infection par des mycobactéries non tuberculeuses, les hémoptysies, l'aspergillose bronchopulmonaire allergique et l'insuffisance cardiaque droite secondaire à l'hypertension pulmonaire.
Gastrointestinal
L'iléus méconial par obstruction de l'iléon par un méconium visqueux peut être le signe le plus précoce; il est présent chez environ 10 à 20% des nouveau-nés atteints de mucoviscidose. Il se manifeste typiquement par une distension abdominale, des vomissements, et une incapacité à éliminer le méconium. Certains nouveau-nés font une perforation intestinale, avec des signes de péritonite et de choc. Les nouveau-nés qui ont un syndrome du bouchon méconial ont un retard d'évacuation du méconium. Ils peuvent avoir des signes semblables d'occlusion intestinale ou des symptômes très bénins et transitoires qui passent inaperçus. Les patients âgés peuvent avoir des épisodes de constipation ou des épisodes récurrents et parfois chroniques d'occlusion partielle ou totale du colon ou du grêle (syndrome d'obstruction intestinale distale). Les symptômes comprennent des crampes abdominales, des changements dans l'apparence des selles, une diminution de l'appétit et parfois des vomissements.
Chez le nourrisson sans iléus méconial, les premières manifestations cliniques peuvent être un retard au rattrapage du poids de naissance et une prise pondérale insuffisante à 4 ou 6 semaines de vie.
Les nourrissons sous-alimentés, en particulier s'ils sont alimentés par des laits hypoallergéniques ou du lait de soja, présentent initialement parfois un œdème généralisé provoqué par la malabsorption des protéines.
L'insuffisance pancréatique est généralement cliniquement apparente tôt au cours de la vie et peut être progressive. Les manifestations cliniques associent des selles fréquentes, huileuses, malodorantes et abondantes; une protubérance abdominale; et un retard de croissance avec diminution du tissu sous-cutané et de la masse musculaire en dépit d'un appétit normal voire augmenté. Certaines manifestations cliniques peuvent être la conséquence d'une carence en vitamines liposolubles.
Un prolapsus rectal peut se produire chez des nourrissons et des enfants en bas âge non traités. Le reflux gastro-œsophagien est relativement fréquent chez l'enfant et l'adulte.
Autres
L'hypersudation lorsque le temps est chaud ou en cas de fièvre peut entraîner des épisodes de déshydratation hyponatrémique/hypochlorémique et une défaillance circulatoire. Dans les climats arides, le nourrisson peut présenter initialement une alcalose métabolique chronique. La présence de sel cristallisé sur la peau ou le goût salé de celle-ci sont très évocateurs d'une mucoviscidose.
Les adolescents peuvent présenter un retard de croissance et un retard pubertaire.
Diagnostic de la mucoviscidose
Dépistage chez le nouveau-né
Peut aussi être suggéré par un dépistage positif prénatal, les antécédents familiaux ou la présentation clinique
Il est confirmé par un test de la sueur montrant une concentration élevée de Cl à 2 reprises
L'identification de 2 variants responsables de la mucoviscidose (1 sur chaque chromosome) est en accord avec le diagnostic
Peut rarement être confirmée, dans les cas atypiques, en démontrant un transport des ions anormal à travers l'épithélium nasal ou des mesures anormales du courant intestinal
La majeure partie des cas des mucoviscidoses sont identifiés par le dépistage néonatal, mais jusqu'à 10% ne sont pas diagnostiqués avant l'adolescence ou l'âge adulte. Malgré les progrès du dépistage génétique, le test de la sueur reste la norme pour confirmer le diagnostic de mucoviscidose dans la plupart des cas, en raison de sa sensibilité et de sa spécificité, de sa simplicité et de sa disponibilité.
Dépistage chez le nouveau-né
Le dépistage universel néonatal de la mucoviscidose est à présent standard aux États-Unis. Le dépistage est basé sur la détection d'une concentration élevée de trypsinogène immunoréactif dans le sang.
Il existe 2 méthodes pour suivre l'évolution d'un taux élevé de trypsinogène immunoréactif. On peut effectuer un second dosage de trypsinogène immunoréactif et s'il est élevé également, il est suivi par un test de la sueur. Dans l'autre méthode plus couramment utilisée, une augmentation du taux de trypsinogène immunoréactif est suivie d'un test de recherche de la mutation du gène CFTR, et si 1 ou 2 variants sont identifiées, alors un test de la sueur est pratiqué. Pour le diagnostic, les deux méthodes ont une sensibilité d'environ entre 90 et 95%.
Test de la sueur
Dans ce test, une transpiration localisée est stimulée par de la pilocarpine, la quantité de sueur est mesurée ainsi que sa concentration en chlore. Bien que la concentration en Cl de la sueur augmente légèrement avec l'âge, le test de la sueur est utilisable à tous les âges:
Normale: ≤ 30 mEq/L (≤ 30 mmol/L) (la mucoviscidose est peu probable.)
Intermédiaire: 30 à 59 mEq/L (30 à 59 mmol/L) (la mucoviscidose est possible)
Anormal: ≥ 60 mEq/L (≥ 60 mmol/L) (ce résultat est compatible avec une mucoviscidose.)
Bien que les résultats du test de la sueur soient valables après 48 heures de vie, un prélèvement suffisant de sueur (> 75 mg sur papier-filtre ou > 15 mcL dans une microtubulure spéciale) peut être difficile à obtenir avant l'âge de 2 semaines. Les faux-négatifs sont rares mais peuvent survenir en cas d'œdèmes et d'hypoprotidémie ou lorsque la quantité de sueur recueillie est insuffisante. Les faux positifs sont habituellement liés à des erreurs techniques. Une augmentation passagère des concentrations de Cl dans la sueur peut être observée en cas de carence psychosociale (p. ex., enfant victime de sévices ou de carences de soins) ou en cas d'anorexie mentale. Un résultat positif au test de la sueur doit être confirmé par un 2e test de la sueur ou par l'identification de 2 variants causes de mucoviscidose.
Résultats intermédiaires au test de la sueur
Certains patients présentant une forme modérée ou partielle de mucoviscidose ont des valeurs de Cl sudoral qui sont de manière persistante dans les valeurs intermédiaires ou même normales. En outre, il existe des patients qui ont des manifestations au niveau d'un seul organe tels qu'une pancréatite chronique ou récurrente, une bronchectasie isolée ou une absence congénitale bilatérale des canaux déférents avec des signes évocateurs de dysfonctionnement de la protéine CFTR. Ils ne répondent pas aux critères diagnostiques de la mucoviscidose et sont classés comme présentant un trouble lié au syndrome CFTR. Chez certains de ces patients, le diagnostic de mucoviscidose peut être confirmé par l'identification des 2 variants causes de mucoviscidose, 1 dans chaque gène chromosome. Si les 2 variantes responsables de mucoviscidose ne sont pas identifiées, des examens complémentaires tels qu'une évaluation de la fonction pancréatique, une imagerie pancréatique et une TDM du thorax à haute résolution, une TDM des sinus, des épreuves fonctionnelles respiratoires, un bilan génito-urinaire chez l'homme, un lavage broncho-alvéolaire et une évaluation de la flore microbienne peuvent être utiles.
D'autres tests diagnostiques tels qu'une analyse extensive du gène CFTR et la mesure des différences de potentiel transépithélial nasal (montrant une augmentation de la réabsorption du sodium à travers l'épithélium qui est relativement imperméable au Cl chez les patients mucoviscidosiques) et la mesure des courants intestinaux peuvent être utiles.
Syndrome métabolique lié au CFTR et diagnostic de mucoviscidose positif, non concluant
Les nourrissons qui présentent un dépistage néonatal positif et des signes d'un possible dysfonctionnement du CFTR mais qui ne répondent pas aux critères de diagnostic de la mucoviscidose sont classés comme ayant un syndrome métabolique lié au CFTR (CFTR-related metabolic syndrome, CRMS), également appelé diagnostic de mucoviscidose positif, diagnostic non concluant (CFSPID). Le diagnostic de syndrome métabolique lié au CFTR (CFTR-related metabolic syndrome, CRMS)/CFSPID (diagnostic de mucoviscidose positif, diagnostic non concluant) est retenu chez les nourrissons qui ont un dépistage néonatal positif, sont asymptomatiques et ont l'une ou l'autre des caractéristiques suivantes:
Les concentrations de chlorure dans la sueur dans la gamme intermédiaire et 0 ou 1 mutation cause de mucoviscidose
Des concentrations normales de chlorure dans la sueur et 2 variants du gène CFTR dont au moins 1 a des conséquences phénotypiques mal définies
La plupart des enfants qui ont un syndrome métabolique lié au CFTR (CFTR-related metabolic syndrome, CRMS)/CFSPID sont en bonne santé, mais sur la durée, environ 10% d'entre eux développeront des symptômes et répondront aux critères diagnostiques de la mucoviscidose ou d'un trouble lié à la mucoviscidose. Les patients qui ont un syndrome métabolique lié au CFTR (CFTR-related metabolic syndrome, CRMS)/CFSPID (diagnostic de mucoviscidose positif, diagnostic non concluant) doivent être évalués et suivis régulièrement dans un centre spécialisé dans la mucoviscidose.
Examens pancréatiques
L'évaluation indirecte et non invasive de la fonction pancréatique peut être réalisée en mesurant généralement la concentration de l'élastase pancréatique humaine dans les selles. La mesure de l'élastase pancréatique humaine est valable même en présence d'enzymes pancréatiques exogènes. Les nourrissons qui ont initialement une fonction pancréatique normale et qui portent 2 variants sévères doivent avoir des mesures répétées pour détecter la progression vers l'insuffisance pancréatique.
Évaluation des voies respiratoires
L'imagerie thoracique est effectuée lors des exacerbations ou en cas d'aggravation de la maladie et systématiquement tous les 1 à 2 ans. La TDM du thorax à haute résolution peut être utile pour définir plus précisément l'étendue des lésions du poumon et détecter des anomalies minimes des voies respiratoires. Les rx et la TDM du thorax peuvent montrer une hyperinflation, une impaction mucoïde et un épaississement des parois bronchiques comme signes les plus précoces. Par la suite apparaissent des infiltrats, des atélectasies et des adénopathies hilaires. À un stade plus avancé, on observe des atélectasies segmentaires ou lobaires, des formations kystiques, des bronchectasies et une augmentation de la taille de l'artère pulmonaire et du ventricule droit. Des opacités ramifiées, en doigt de gant, correspondant à des bouchons de mucus dans des bronches dilatées, sont caractéristiques.
La TDM des sinus est indiquée en cas de symptômes sinusiens ou de polypes nasaux si une chirurgie endoscopique des sinus est envisagée. Ces examens montrent presque toujours une opacification persistante des sinus de la face.
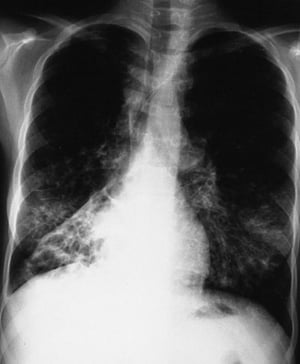
By permission of the publisher. D'après Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.
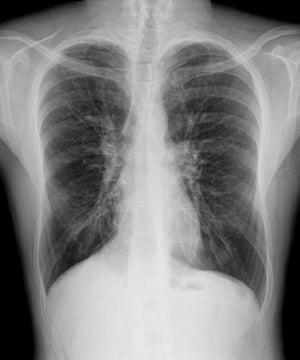
PHOTOSTOCK-ISRAEL/SCIENCE PHOTO LIBRARY
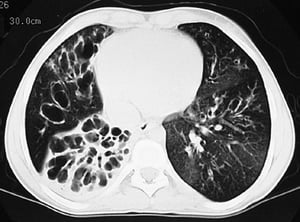
By permission of the publisher. D'après Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.

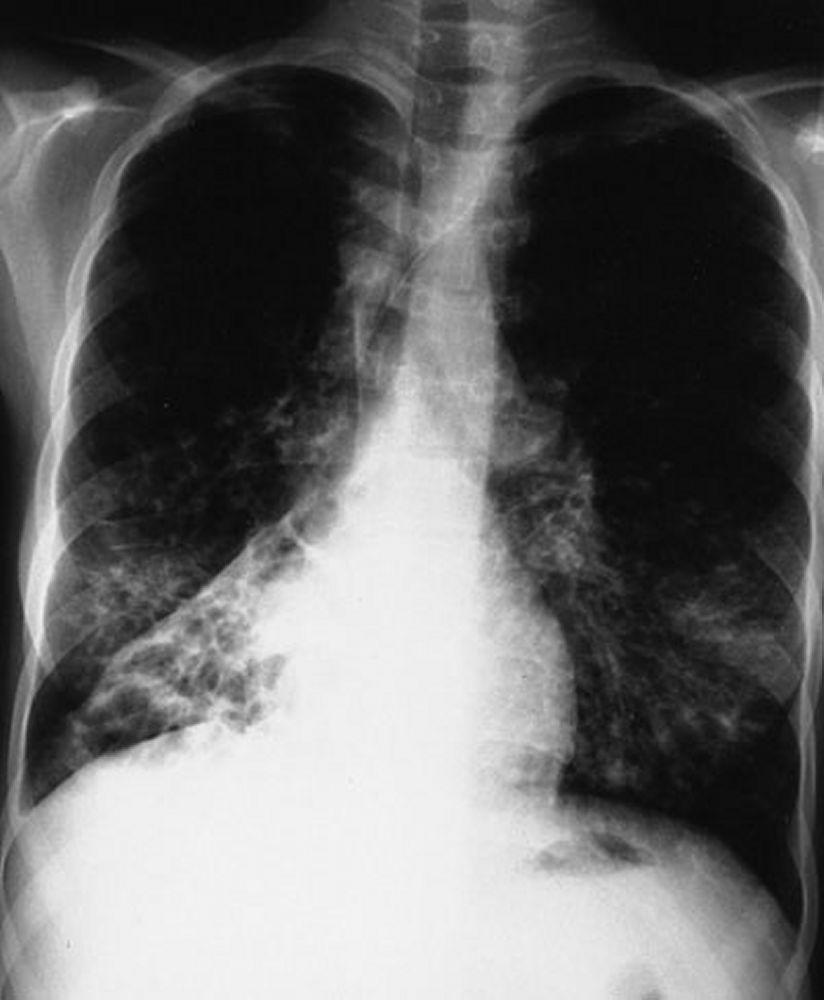
By permission of the publisher. D'après Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.

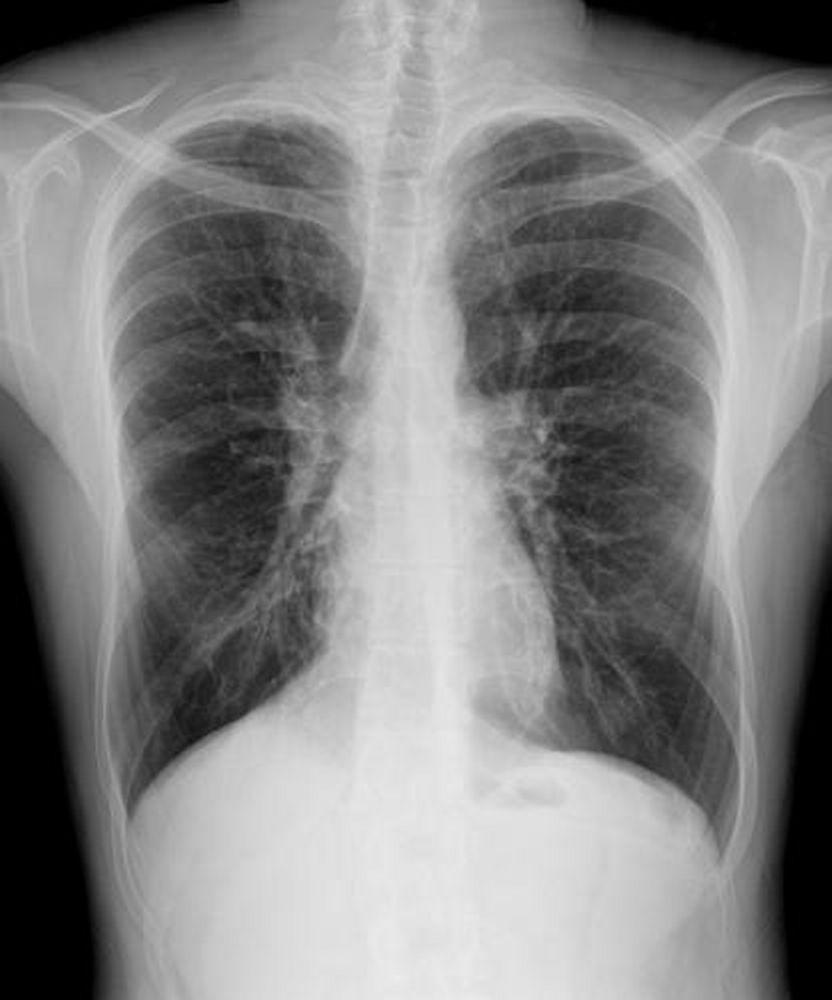
PHOTOSTOCK-ISRAEL/SCIENCE PHOTO LIBRARY

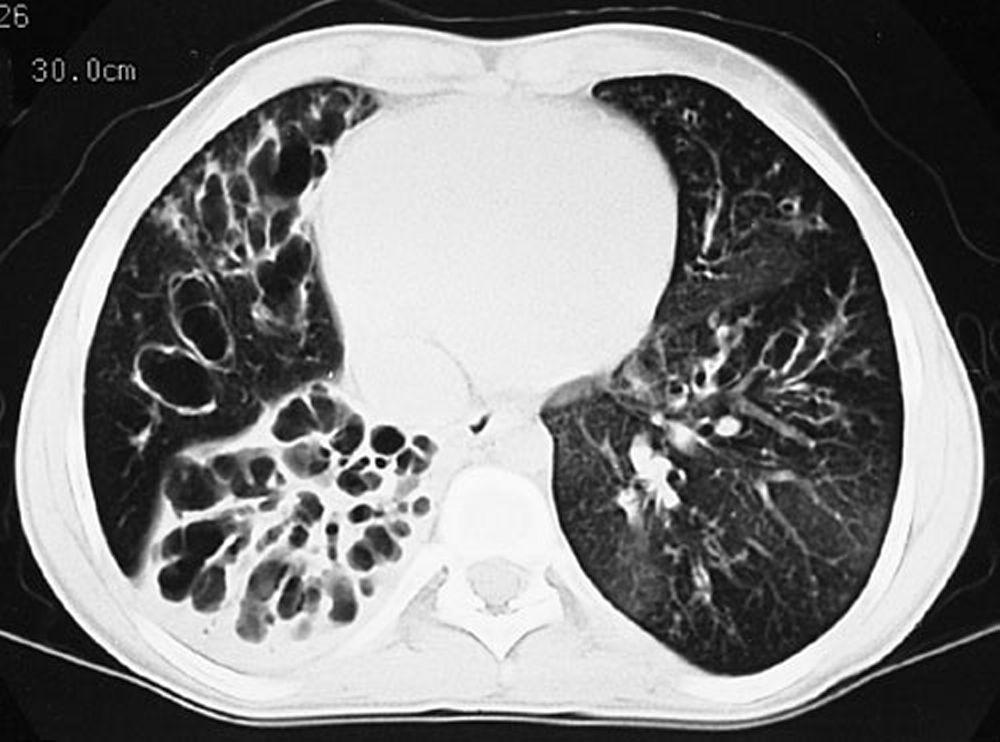
By permission of the publisher. D'après Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.
Les épreuves fonctionnelles respiratoires sont les meilleurs indicateurs de l'état clinique et de la réponse au traitement. Chez les patients de plus de 5 ans d'âge, la spirométrie doit être effectuée systématiquement et en cas de déclin clinique. Chez les nourrissons, l'état respiratoire peut être surveillé en utilisant une technique de compression thoracoabdominale rapide à volume élevé, qui génère une courbe débit-volume partielle. Chez l'enfant de 3 à 6 ans, la procédure de sevrage respiratoire multiple peut être utilisée pour générer un indice de clairance pulmonaire comme mesure de l'inhomogénéité de la ventilation (1).
Les preuves fonctionnelles respiratoires effectuées par spirométrie montrent
Une réduction de la capacité vitale forcée (CVF), volume expiratoire maximum en 1 s (VEMS1), le débit expiratoire forcé entre 25% et 75% du volume expiré (FEF25-75) et le rapport VEMS1/CVF
Une augmentation du volume résiduel et le rapport du volume résiduel sur la capacité pulmonaire totale
Cinquante pour cent des patients présentent des éléments en faveur d'une obstruction réversible des voies respiratoires, démontrée par l'amélioration de la fonction respiratoire après administration de bronchodilatateurs inhalés.
Des cultures de l'expectoration ou des sécrétions oropharyngées doivent être effectuées au moins 4 fois/an, en particulier chez les patients non encore colonisés par P. aeruginosa. Une bronchoscopie/lavage bronchoalvéolaire est indiquée quand il est important de définir avec précision la flore microbienne des voies respiratoires du patient (p. ex., pour orienter le choix des antibiotiques) ou pour éliminer des bouchons muqueux épaissis.
Dépistage des porteurs (hétérozygotes)
Le dépistage des hétérozygotes pour la mucoviscidose (porteurs asymptomatiques de la mutation) est disponible aux États-Unis et il est recommandé pour les couples qui planifient une grossesse ou qui recherchent des soins prénatals. Si les deux parents potentiels sont porteurs d'un variant de CFTR, un dépistage prénatal du fœtus peut être effectué par prélèvement de villosités choriales ou par amniocentèse. Le conseil prénatal dans de tels cas est compliqué par la grande variabilité phénotypique de la mucoviscidose et les informations incomplètes sur les conséquences cliniques de nombre de variants du gène CFTR qui sont identifiées par un dépistage.
Référence pour le diagnostic
1. Stanojevic S, Davis SD, Retsch-Bogart G, et al: Progression of lung disease in preschool patients with cystic fibrosis. Am J Respir Crit Care Med 195:1216–1225, 2017. doi: 10.1164/rccm.201610-2158OC
Traitement de la mucoviscidose
Adapté au patient, avec un soutien pluridisciplinaire
Antibiotiques, médicaments inhalés pour fluidifier les sécrétions et des techniques physiques de désencombrement des voies
Inhalations de bronchodilatateurs et parfois des corticostéroïdes pour ceux qui y répondent
Habituellement supplémentation en enzymes pancréatiques et en vitamines
Régime à haute teneur en calories (nécessitant parfois un tube entérique)
Chez les patients présentant des variants spécifiques, les modulateurs de CFTR constitués d'un potentialisateur CFTR ou d'une association de correcteurs CFTR et d'un potentiateur CFTR
Le traitement intensif et global doit être conduit par un praticien expérimenté travaillant avec une équipe multidisciplinaire qui comprend d'autres médecins, des infirmières, des nutritionnistes, des kinésithérapeutes, des professionnels de la santé mentale, des pharmaciens et des assistants sociaux. Les objectifs thérapeutiques sont le maintien d'un bon état nutritionnel, le traitement préventif ou curatif des complications pulmonaires et des autres complications, l'encouragement à une activité physique et la mise en place d'un soutien psychosocial adapté. Le protocole thérapeutique est complexe et peut prendre jusqu'à 2 heures par jour. Avec toutes les aides adéquates, la plupart des patients peuvent avoir chez eux comme à l'école des activités correspondant à leur âge.
(Voir aussi the Cystic Fibrosis Foundation's comprehensive treatment guidelines for all age groups.)
Traitement des manifestations respiratoires
Le traitement des manifestations pulmonaires est centré sur la prévention de l'obstruction des voies respiratoires et la surveillance des infections pulmonaires. La prévention des infections respiratoires comprend le maintien de l'immunité vaccinale vis-à-vis de la coqueluche, d'Haemophilus influenzae, de la varicelle, de Streptococcus pneumoniae, de la rougeole et la vaccination antigrippale refaite annuellement; et la vaccination contre le COVID-19 conformément aux recommandations actuelles de l'Advisory Committee on Immunization Practices (ACIP). Chez le patient exposé à la grippe, un inhibiteur de neuraminidase peut être utilisé préventivement ou aux premiers signes d'infection. L'administration du nirsevimab, ou lorsqu'il n'est pas disponible, du palivizumab chez les nourrissons qui présentent une mucoviscidose, pour la prévention de l'infection par le virus respiratoire syncytial a été démontrée être sans danger, mais son efficacité n'a pas été prouvée.
La thérapie d'inhalation quotidienne à long terme par l'alphadornase (désoxyribonucléase recombinante humaine) ou par une solution saline hypertonique à 7% est recommandée (1, 2) et a été démontrée ralentir la vitesse du déclin de la fonction pulmonaire et diminuer la fréquence des exacerbations respiratoires (3).
Les mesures de drainage des voies respiratoires consistant en un drainage postural, une percussion, une vibration et une toux assistée (kinésithérapie assistée) sont recommandées au moment du diagnostic et doivent être effectuées de façon régulière. Chez le patient âgé, d'autres mesures de drainage des voies respiratoires, telles que le cycle de respiration active, le drainage autogène, des dispositifs de pression expiratoire positive et le traitement par gilet (oscillations à haute fréquence de la paroi thoracique) peuvent être efficaces. Un exercice aérobie régulier est recommandé; il peut aussi permettre de dégager les voies respiratoires. En cas d'apnée obstructive du sommeil, la pression positive continue des voies respiratoires peut être bénéfique.
En cas de signes d'obstruction des voies respiratoires, des bronchodilatateurs peuvent être administrés par inhalation. Les corticostéroïdes en inhalation sont habituellement efficaces. L'oxygénothérapie est indiquée chez le patient présentant une insuffisance respiratoire sévère avec hypoxémie.
La ventilation mécanique ou l'oxygénation par membrane extracorporelle (ECMO) n'est généralement pas indiquée dans l'insuffisance respiratoire chronique. Leur utilisation est généralement réservé au patient en état basal satisfaisant chez qui une complication respiratoire aiguë réversible apparaît, en association avec une chirurgie pulmonaire ou chez le patient chez lequel une transplantation pulmonaire est imminente. La ventilation non invasive en pression positive, par masque nasal ou facial, peut également être utile.
Les expectorants par voie orale sont parfois utilisés, mais peu de données attestent de leur efficacité. Les médicaments antitussifs doivent être déconseillés.
Le pneumothorax peut être traité par drainage thoracique percutané par cathéter de thoracostomie. La thoracotomie à ciel ouvert ou la thoracoscopie pour résection de bulles sous-pleurales et abrasion mécanique des surfaces pleurales est efficace dans le traitement des pneumothorax récidivants.
L'hémoptysie légère à modérée est traitée par des antibiotiques (par voie orale/aérosol ou IV selon la gravité de l'hémoptysie et la gravité de l'infection) et l'évacuation des voies respiratoires. L'hémoptysie massive ou récurrente est traitée par embolisation de l'artère bronchique ou rarement par résection pulmonaire focale.
Les corticostéroïdes par voie orale sont indiqués chez le nourrisson souffrant d'une bronchiolite prolongée et chez le patient présentant un bronchospasme réfractaire avec aspergillose bronchopulmonaire allergique et en cas de complications inflammatoires (p. ex., arthrites et vascularites). L'utilisation à long terme des corticostéroïdes en traitement 1 jour/2 peut ralentir la détérioration de la fonction respiratoire, mais, du fait des complications propres des corticostéroïdes, ils ne sont pas recommandés de manière systématique. Le patient recevant des corticostéroïdes doit être étroitement surveillé afin de repérer l'apparition d'anomalies du métabolisme glucidique ou d'un retard de croissance staturale.
L'aspergillose bronchopulmonaire allergique est également traitée par des corticostéroïdes systémiques et un antifongique oral.
L'ibuprofène, administré pendant plusieurs années, à une dose suffisante pour obtenir une concentration plasmatique de pic entre 50 et 100 mcg/mL (242,4 et 484,8 micromoles/L), a été montré ralentir la vitesse de déclin de la fonction pulmonaire, en particulier chez l'enfant de 5 à 13 ans. La posologie appropriée doit être individualisée en se basant sur les études pharmacocinétiques.
La rhinosinusite chronique est très fréquente. Les options de traitement comprennent l'irrigation nasale avec une solution physiologique, l'irrigation nasale isotonique à basse pression, la nébulisation intranasale de dornase alpha et des antibiotiques topiques nasaux. La chirurgie des sinus peut être utile dans les cas réfractaires à la prise en charge médicale. Une pulvérisation intranasale de corticostéroïdes est recommandée pour traiter la rhinite allergique.
Modulateurs de CFTR
Les médicaments correcteurs et potentialisateurs du CFTR sont indiqués dans le cas d'environ 90% des variants portés par les patients qui ont une mucoviscidose. Les modulateurs du CFTR ne sont pas disponibles chez les patients présentant des mutations de classe I et non-sens.
L'ivacaftor est un médicament petite molécule qui potentialise le canal ionique CFTR chez les patients présentant des variantes spécifiques de CFTR. Il peut être utilisé chez les patients de 1 mois d'âge et plus qui sont porteurs d'au moins 1 copie d'un variant spécifique potentialisé par l'ivacaftor.
Le lumacaftor, le tézacaftor et l'élexacaftor sont des médicaments petite molécule qui corrigent partiellement la protéine CFTR défectueuse en modifiant le repliement anormal des protéines chez les patients porteurs du variant F508del et d'autres variants spécifiques.
L'association de lumacaftor et d'ivacaftor peut être administrée aux patients de 1 an et plus porteurs de 2 copies du variant F508del.
L'association de tezacaftor et d'ivacaftor peut être administrée aux patients de 6 ans et plus porteurs de 2 copies du variant F508del ou d'autres variants spécifiques.
L'association triple d'elexacaftor, de tezacaftor et d'ivacaftor peut être administrée aux patients âgés de 2 ans et plus qui ont au moins 1 copie de la variante F508del ou 1 copie de certaines variantes rares (4, 5).
Ces médicaments peuvent améliorer la fonction pulmonaire, augmenter le poids, améliorer la fonction pancréatique exocrine, diminuer la fréquence des exacerbations pulmonaires et des hospitalisations, améliorer la qualité de vie et réduire et parfois normaliser les concentrations de chlorure dans la sueur (6). Les indications de l'ivacaftor, du lumacaftor/ivacaftor, du tezacaftor/ivacaftor et de l'elexacaftor/tezacaftor/ivacaftor sont basées sur les variants CFTR du patient et son âge et elles évoluent rapidement. Bien que tous ces médicaments puissent être utiles, seuls l'ivacaftor et l'association d'elexacaftor, de tezacaftor et d'ivacaftor sont considérés comme des modulateurs très efficaces.
Traitement et prévention des infections
En cas d'exacerbations pulmonaires bénignes, une courte cure d'antibiotiques oraux doit être administrée en fonction de la culture et des antibiogrammes. Les médicaments de choix pour les staphylocoques sensibles à la méthicilline sont une pénicilline résistante à la pénicillinase (p. ex., dicloxacilline), une céphalosporine (p. ex., céphalexine) ou le triméthoprime/sulfaméthoxazole. L'érythromycine, l'amoxicilline/clavulanate, la tétracycline ou le linézolide peuvent être utilisés. Chez les patients colonisés par S. aureus résistant à la méthicilline (SARM), un traitement par le triméthoprime/sulfaméthoxazole, la clindamycine, le linézolide ou une tétracycline peuvent être efficaces. Dans le cas de patients colonisés par P. aeruginosa, un bref traitement par la tobramycine ou l'aztréonam inhalés (p. ex., 4 semaines) et/ou une fluoroquinolone orale (p. ex., de 2 à 3 semaines) peut être efficace. Les fluoroquinolones ont été utilisées en toute sécurité chez l'enfant.
En cas d'exacerbations pulmonaires modérées à sévères, en particulier chez le patient colonisé par P. aeruginosa, une antibiothérapie IV est conseillée. Les patients ont souvent besoin d'être hospitalisés, mais des patients soigneusement sélectionnés peuvent recevoir sans danger une partie du traitement à domicile. Des associations de l'aminoside tobramycine, ou parfois de l'amikacine et d'une céphalosporine, de pénicilline à spectre étendu, de fluoroquinolone ou d'un monobactame à activité antipseudomonale sont administrées par voie IV, généralement pendant 2 semaines. Des doses plus élevées peuvent être nécessaires pour atteindre des concentrations sériques acceptables. La clairance rénale étant augmentée chez les patients qui souffrent de mucoviscidose, de fortes doses de certaines pénicillines peuvent être nécessaires pour atteindre des concentrations plasmatiques suffisantes. Dans le cas de patients colonisés par un S. aureus, résistant à la méthicilline, la vancomycine ou le linézolide peuvent être ajoutés au traitement IV.
L'éradication de la colonisation chronique par P. aeruginosa est difficile. Cependant, il a été démontré qu'un traitement antibiotique précoce, débutant environ au moment où les voies respiratoires sont initialement infectées par P. aeruginosa, peut s'avérer efficace et permettre d'éradiquer ce microrganisme pendant un certain temps. Chez le patient chroniquement colonisé par P. aeruginosa, les antibiotiques administrés par inhalation améliorent les paramètres cliniques et peuvent réduire la charge bactérienne dans les voies respiratoires (7). L'administration à long terme avec un mois sur deux de tobramycine en inhalation ou d'aztréonam avec de l'azithromycine par voie orale en continu (chaque mois) administrée 3 fois/semaine peut s'avérer efficace pour améliorer ou stabiliser la fonction respiratoire et diminuer la fréquence des épisodes d'aggravations pulmonaires.
Les patients qui ont une infection à mycobactérie non tuberculeuse cliniquement significative peuvent nécessiter un traitement à long terme par une association d'antibiotiques oraux, inhalés, et IV.
L'aspergillose broncho-pulmonaire allergique ou les infections des voies respiratoires inférieures par aspergillus peuvent nécessiter un traitement prolongé par voie orale ou intraveineuse avec un azole antifongique et/ou des corticostéroïdes systémiques.
Traitement des manifestations digestives
L'occlusion intestinale néonatale peut parfois être levée par des lavements contenant un produit de contraste radio-opaque hyper- ou iso-osmolaire; dans d'autres cas, une entérostomie chirurgicale pour évacuer le méconium visqueux dans la lumière intestinale peut être nécessaire. Après la période néonatale, les épisodes d'occlusion intestinale partielle (syndrome d'occlusion intestinale distale) peuvent être traités par des lavements similaires ou contenant de l'acétylcystéïne ou par l'administration orale d'une solution équilibrée de lavement intestinal. Un émollient tel que le dioctylsulfosuccinate de sodium (docusate) ou le lactulose permet de prévenir ces épisodes.
L'acide ursodésoxycholique, un acide biliaire hydrophile, est souvent utilisé en cas d'atteinte hépatique causée par la mucoviscidose, mais il existe peu de preuves en faveur de son efficacité pour empêcher la stase biliaire d'évoluer en cirrhose.
Les enzymes pancréatiques de substitution doivent être administrées à tous les repas et toutes les collations chez le patient présentant une insuffisance pancréatique. Les préparations enzymatiques les plus efficaces contiennent de la pancrélipase enrobée dans des microsphères ou des microcomprimés sensibles au pH, avec libération intestinale. Pour les nourrissons, les gélules sont ouvertes et les contenus sont mélangés avec de la nourriture acide. Après la petite enfance, un dosage en fonction du poids est utilisé. Les doses > 2500 UI de lipase/kg/repas ou > 10 000 UI de lipase/kg/jour doivent être évitées, car des posologies élevées d'enzyme ont été associées à la survenue d'une colopathie fibrosante. Chez le patient ayant des besoins élevés en enzymes, l'utilisation d'un anti-H2 ou d'un inhibiteur de la pompe à protons peut améliorer l'efficacité de l'enzyme.
Le régime comprend des calories et des protéines suffisantes pour promouvoir une croissance normale, 30 à 50% de plus que les apports nutritionnels conseillés habituels peuvent être nécessaires (voir tableau Apports nutritionnels recommandés pour certains macronutriments). Le régime comprend également des graisses en quantité de normale à élevée pour augmenter la densité calorique du régime, des suppléments multivitaminiques miscibles à l'eau en quantité double de l'apport quotidien recommandé, une supplémentation en vitamine D3 (cholécalciférol) en cas de déficit en vitamine D; et une supplémentation en sel au cours de la petite enfance et des périodes de stress thermique et de transpiration augmentée. Le nourrisson traité par des antibiotiques à large spectre et le patient qui a une atteinte hépatique ou des hémoptysies doivent recevoir un supplément de vitamine K. Des préparations artificielles particulières, contenant des hydrolysats de protéines et des triglycérides à chaîne moyenne, peuvent être utilisées au lieu du lait artificiel entier chez le nourrisson en cas de malabsorption sévère. Des suppléments de polymères de glucose et de triglycérides à chaîne moyenne peuvent être utilisés afin d'augmenter l'apport calorique.
Chez les patients chez qui l'on a des difficultés à maintenir un bon état nutritionnel, une supplémentation entérale par gastrostomie ou jéjunostomie peut améliorer la croissance et stabiliser la fonction pulmonaire (voir Revue générale des apports nutritionnels). L'utilisation de stimulants d'appétit pour améliorer la croissance peut être utile chez certains patients.
Traitement d'autres manifestations
Le diabète lié à la mucoviscidose est causé par une carence en insuline et partage des caractéristiques du diabète de type 1 et 2. L'insuline est le seul traitement recommandé. La prise en charge comprend un protocole insulinique, des conseils nutritionnels, un programme d'enseignement de l'autogestion du diabète, et de surveillance des complications microvasculaires. Le plan doit être exécuté en conjonction avec un endocrinologue et un diététicien qui a l'expérience du traitement de la mucoviscidose et du diabète.
Le patient présentant une insuffisance cardiaque droite symptomatique doit être traité par diurétiques, restriction sodée et oxygène.
L'hormone de croissance humaine recombinante (rhGH) peut améliorer la fonction pulmonaire, augmenter la taille et le poids et la teneur minérale des os, et réduire les taux d'hospitalisation. Cependant, en raison de son coût et des inconvénients supplémentaires, la rhGH n'est pas couramment utilisée.
La chirurgie peut être indiquée en cas de bronchectasies ou d'atélectasies localisées ne pouvant être traitées efficacement par le traitement médicamenteux, en cas de polypes nasaux, de rhinosinusite chronique, d'hématémèses dues à des varices œsophagiennes secondaires à une hypertension portale, de pathologie de la vésicule biliaire ou d'occlusion intestinale par volvulus ou invagination ne pouvant être réduite médicalement.
Des transplantations hépatiques ont été accomplies avec succès en cas d'hépatopathie parvenue à un stade terminal.
Souvent, une discussion à propos de la transplantation pulmonaire est nécessaire. En ce qui concerne la transplantation, le patient doit évaluer les avantages d'une survie plus longue grâce à la greffe, mais avec l'incertitude d'obtenir le greffon et de vivre avec une maladie (bien que différente) liée au fait de vivre avec un organe transplanté. La transplantation bilatérale de poumon de cadavre et lobaire par donneur vivant a été réalisée avec succès chez des patients qui ont une maladie pulmonaire avancée. Une transplantation combinée foie-poumon a été effectuée chez des patients qui ont une maladie hépatique et pulmonaire en phase terminale.
La transplantation pulmonaire bilatérale en cas de maladie pulmonaire sévère est de plus en plus courante avec des chances de succès améliorées par l'expérience et les progrès techniques. Chez les adultes atteints de mucoviscidose, la survie médiane après greffe est d'environ 9 ans.
Références pour le traitement
1. Flume PA, O'Sullivan BP, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007;176(10):957-969. doi:10.1164/rccm.200705-664OC
2. Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2013;187(7):680-689. doi:10.1164/rccm.201207-1160oe
3. Stahl M, Wielpütz MO, Ricklefs I, et al. Preventive Inhalation of Hypertonic Saline in Infants with Cystic Fibrosis (PRESIS). A Randomized, Double-Blind, Controlled Study. Am J Respir Crit Care Med 2019;199(10):1238-1248. doi:10.1164/rccm.201807-1203OC
4. Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial [published correction appears in Lancet 2020 May 30;395(10238):1694]. Lancet 2019;394(10212):1940-1948. doi:10.1016/S0140-6736(19)32597-8
5. Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 2019;381(19):1809-1819. doi:10.1056/NEJMoa1908639
6. Taylor-Cousar JL, Robinson PD, Shteinberg M, Downey DG. CFTR modulator therapy: transforming the landscape of clinical care in cystic fibrosis. Lancet 2023;402(10408):1171-1184. doi:10.1016/S0140-6736(23)01609-4
7. Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic Fibrosis Foundation pulmonary guideline. pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann Am Thorac Soc 2014;11(10):1640-1650. doi:10.1513/AnnalsATS.201404-166OC
Pronostic de la mucoviscidose
L'évolution est largement déterminée par la gravité de l'atteinte pulmonaire. La détérioration de la fonction pulmonaire au fil du temps, généralement caractérisée par une bronchectasie progressive, conduit à un débilitation et finalement augmente le risque de décès, habituellement dû à une association d'insuffisance respiratoire et de cœur pulmonaire.
Le pronostic s'est progressivement amélioré au cours des 5 dernières décennies, grâce principalement à un diagnostic précoce et à l'instauration de traitements intensifs avant l'apparition de lésions pulmonaires irréversibles. L'âge médian au décès en 2021 était de 33,9 ans. Cependant, la médiane de survie aux États-Unis des enfants nés en 2021 est de 65,6 ans. La survie à long terme est significativement plus longue en l'absence d'insuffisance pancréatique (1). Le pronostic est également influencé par le profil des variants CFTR, par les gènes modificateurs, la microbiologie des voies respiratoires, le sexe, la température ambiante, l'exposition aux polluants de l'air (notamment la fumée de tabac), le respect des traitements prescrits et le statut socio-économique. Le VEMS1, corrigé pour l'âge et le sexe, est le meilleur facteur prédictif de la mortalité. Si les résultats de santé du traitement par modulateur du CFTR sont durables, l'espérance de vie peut potentiellement augmenter encore plus.
Soins de fin de vie
Les patients et leurs familles méritent des discussions empreintes de sensibilité en ce qui concerne le pronostic et les préférences de soins tout au long de l'évolution de la maladie, en particulier si la fonction pulmonaire diminue progressivement.
Une marque de respect à l'égard d'une personne qui vit avec la mucoviscidose est d'être certain qu'on lui fournit les informations et les possibilités de faire ses choix, y compris sur le fait d'avoir la possibilité de pouvoir décider comment et quand accepter de mourir.
Le cas échéant, des soins palliatifs, dont une prise en charge suffisante des symptômes, doivent être proposés pour assurer des soins de fin de vie pacifiés. Une stratégie utile à envisager par le patient est d'accepter l'essai sur une durée limitée d'un traitement très agressif si nécessaire, mais d'accepter à l'avance les paramètres qui indiqueront le moment où ces mesures agressives devront être arrêtées (voir Instructions de "ne pas réanimer" et instructions de médecins pour les traitements de réanimation [Do-Not-Resuscitate (DNR) Orders and Portable Medical Orders).
Référence pour le pronostic
1. Cystic Fibrosis Foundation Patient Registry 2021 Annual Data Report Bethesda, Maryland 2022 Cystic Fibrosis Foundation. Consulté le 20 octobre 2023.
Points clés
La mucoviscidose est provoquée par la présence de 2 variants du gène codant pour une protéine appelée le régulateur de conductance transmembranaire de la mucoviscidose (CFTR, cystic fibrosis transmembrane conductance regulator), qui régule le transport du sodium, du chlore et du bicarbonate à travers les membranes épithéliales.
Les principales complications touchent les poumons, avec des lésions des petites et des grandes voies respiratoires, une inflammation chronique, et des infections bactériennes chroniques et récurrentes, notamment par Pseudomonas aeruginosa.
D'autres conséquences majeures comprennent un insuffisance pancréatique, induisant une malabsorption des nutriments et des vitamines avec comme conséquence un trouble de la croissance et du développement, et, chez les patients âgés, un risque de diabète.
Les mesures de dégagement des voies respiratoires (p. ex., drainage postural, percussion, vibration, toux assistée), les mucolytiques et les hydrateurs des voies respiratoires sont souvent commencés dans la petite enfance; des exercices aérobies réguliers sont recommandés.
Les médicaments qui corrigent ou potentialisent le CFTR (modulateurs du CFTR) peuvent améliorer l'évolution des patients qui ont certaines variantes du CFTR.
Les antibiotiques sont administrés au début de toute exacerbation pulmonaire; la sélection des médicaments peut être fonction de la culture et des antibiogrammes.
L'alimentation doit être complétée par des enzymes pancréatiques, des vitamines à haute dose et 30 à 50% de plus de calories provenant principalement de la graisse.
Plus d'information
La source d'information suivante en anglais peut être utile. S'il vous plaît, notez que LE MANUEL n'est pas responsable du contenu de cette ressource.
Cystic Fibrosis Foundation: Age-specific care guidelines for cystic fibrosis







